|
|
4-phenylbutyric Acid sodium salt
4-苯基丁酸鈉鹽
https://en.wikipedia.org/wiki/Sodium_phenylbutyrate
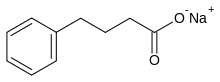
Sodium phenylbutyrate is a salt of an
aromatic fatty acid, 4-phenylbutyrate (4-PBA) or 4-phenylbutyric acid.[1] The
compound is used to treat urea cycle disorders, because its metabolites offer an
alternative pathway to the urea cycle to allow excretion of excess
nitrogen.[2][3] It is an orphan drug, marketed by Ucyclyd Pharma under the trade
name Buphenyl, by Swedish Orphan International (Sweden) as Ammonaps, and by
Fyrlklövern Scandinavia as triButyrate.
Sodium phenylbutyrate is also a histone
deacetylase inhibitor and chemical chaperone, leading respectively to research
into its use as an anti-cancer agent and in protein misfolding diseases such as
cystic fibrosis.[1] abc
Acyclovir
阿昔洛韋
https://en.wikipedia.org/wiki/Aciclovir
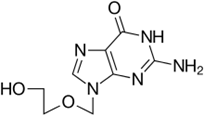
Aciclovir (ACV), also known as acyclovir, is
an antiviral medication.[3] It is primarily used for the treatment of herpes
simplex virus infections, chickenpox, and shingles. Other uses include
prevention of cytomegalovirus infections following transplant and infections due
to Epstein-Barr virus. It is available by mouth and intravenously.[4]
Common side effects include nausea and
diarrhea. Potentially serious side effects include kidney problems and low
platelets. Greater care is recommended in those with poor liver or kidney
function.[4] It is generally considered safe for use inpregnancy with no harm
having been observed.[4][5] It appears to be safe during breastfeeding.[6][7]
Aciclovir is a nucleic acid analogue made from guanosine. It works by decreasing
the production of the virus's DNA.[4]
The discovery of aciclovir was announced in
1977.[8] It is on the World Health Organization's List of Essential Medicines,
the most important medications needed in a basic health system.[9] It is
available as a generic medication and is marketed under many brand names
worldwide.[1] The wholesale cost as of 2014 to 2016 was between US$0.03 and
US$0.12 for a typical dose by mouth.[10][11] The cost of a typical course of
treatment in the United States is less than US$25.[6]
Alfacalcidol
阿法骨化醇
https://en.wikipedia.org/wiki/Alfacalcidol
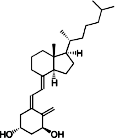
Alfacalcidol (or 1-hydroxycholecalciferol)
is an analogue of vitamin D used for supplementation in humans and as a poultry
feed additive.
Alfacalcidol has a weaker impact on calcium
metabolism[1] and parathyroid hormone levels[2] than calcitriol, however
alfacalcidol has significant effects on the immune system, including regulatory
T cells.[3] It is considered to be a more useful form of vitamin D
supplementation, mostly due to much longer half-life and lower kidney load.[4]
It is the most commonly prescribed vitamin D metabolite for patients with end
stage renal disease, given that impaired renal function alters the ability to
carry out the second hydroxylation step required for the formation of the
physiologically active form of vitamin D, 1,25-dihydroxyvitamin D3. Alfacalcidol
is an active vitamin D3 metabolite, and therefore does not require the
secondhydroxylation step in the kidney.[5]
Used as a poultry feed additive, it prevents
tibial dyschondroplasia and increases phytate bioavailability.[6][original
research?]
Alfuzosin Hydrochloride
阿福唑嗪
https://en.wikipedia.org/wiki/Alfuzosin
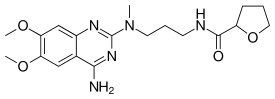
Alfuzosin (INN, provided as the
hydrochloride salt) is a pharmaceutical drug of the α1 blocker class. As an
antagonist of the α1 adrenergic receptor, it works by relaxing the muscles in
the prostate and bladder neck, making it easier to urinate. It is thus used to
treat benign prostatic hyperplasia (BPH).
Alfuzosin is marketed in the United States
by Sanofi Aventis under the brand name Uroxatral and elsewhere under the
tradenames Xat, Xatral, Prostetrol and Alfural. Alfuzosin was approved by the
U.S. FDA for treatment of BPH in June 2003.
Allopurinol
別嘌呤醇
https://en.wikipedia.org/wiki/Allopurinol
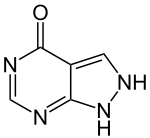
Allopurinol, sold under the brand name
Zyloprim and generics, is a medication used primarily to treat excess uric acid
in the blood and its complications, including chronic gout.[1] It is a xanthine
oxidase inhibitor and is administered orally.
It is on the World Health Organization's
List of Essential Medicines, a list of the most important medication needed in a
basic health system.[2]
Alprazolam
阿普唑侖
https://en.wikipedia.org/wiki/Alprazolam
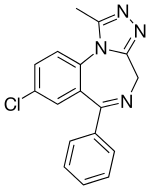
Alprazolam, available under the trade name
Xanax (and sometimes known as xans or zans for short) is a
short-actinganxiolytic of the benzodiazepine class. It is commonly used for the
treatment of panic disorder, and anxiety disorders, such as generalized anxiety
disorder (GAD) or social anxiety disorder (SAD).[4][5] It was the 12th most
prescribed medicine in the USA in 2010.[6] Alprazolam, like other
benzodiazepines, binds to specific sites on the GABAA receptor. It
possessesanxiolytic, sedative, hypnotic, skeletal muscle relaxant,
anticonvulsant, and amnestic properties.[7] Alprazolam is available fororal
administration in compressed tablet (CT) and extended-release capsule (XR)
formulations.
Alprostadil
前列地爾
https://en.wikipedia.org/wiki/Prostaglandin_E1
Prostaglandin E1 (PGE1) is a prostaglandin.
The synthetic variant is known
pharmaceutically as alprostadil.[1] It is a drug used in the continuous
treatment of erectile dysfunction[2] and has vasodilatory properties.
Misoprostol is another synthetic prostaglandin E1 analog used to prevent gastric
ulcers when taken on a continuous basis, to treat missed miscarriage, to induce
labor, and to induce abortion.
Anastrozole
阿那曲唑
https://en.wikipedia.org/wiki/Anastrozole
Aromatase inhibitors (AIs) are a class of
drugs used in the treatment of breast cancer in postmenopausal women
andgynecomastia in men. They may also be used off-label to reduce increase of
estrogen conversion during cycle with external testosterone. They may also be
used for chemoprevention in high risk women.
Aromatase is the enzyme that synthesizes
estrogen. As breast and ovarian cancers require estrogen to grow, AIs are taken
to either block the production of estrogen or block the action of estrogen on
receptors.
Argatroban
阿加曲班
https://en.wikipedia.org/wiki/Argatroban
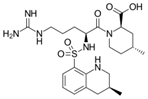
Argatroban is an anticoagulant that is a
small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by
theFood and Drug Administration (FDA) for prophylaxis or treatment of thrombosis
in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was
approved for use during percutaneous coronary interventions in patients who have
HIT or are at risk for developing it. In 2012, it was approved by the MHRA in
the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia
Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug
plasma concentrations reach steady state in 1–3 hours.[3] Argatroban is
metabolized in the liver and has a half-life of about 50 minutes. It is
monitored by PTT. Because of its hepatic metabolism, it may be used in patients
with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin
inhibitor that is primarily renally cleared).
Articaine Hydrochloride
鹽酸阿尼卡因
https://en.wikipedia.org/wiki/Articaine
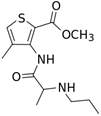
Articaine is a dental amide-type local
anesthetic. It is the most widely used local anesthetic in a number of European
countries[2] and is available in many countries around.
Atenolol
阿替洛爾
https://en.wikipedia.org/wiki/Atenolol
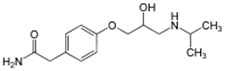
Atenolol is a selective β1 receptor
antagonist, a drug belonging to the group of beta blockers (sometimes written
β-blockers), a class of drugs used primarily in cardiovascular diseases.
Introduced in 1976, atenolol was developed as a replacement for propranolol in
the treatment of hypertension. It works by slowing down the heart and reducing
its workload. Unlike propranolol, atenolol does not readily pass through the
blood–brain barrier, thus decreasing the incidence of central nervous system
side effects.[1]
Atenolol is one of the most widely used
β-blockers in the United Kingdom and was once the first-line treatment for
hypertension.[citation needed] However, recent studies indicate that atenolol
may not reduce morbidity or mortality when used to treat hypertension, and may
even increase mortality in some subgroups.[2] In addition, the role for
β-blockers in general in hypertension was downgraded in June 2006 in the United
Kingdom, and later in the United States, as they are less appropriate than newer
drugs, particularly in the elderly.[citation needed]
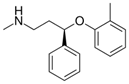
Atomoxetine HCl
阿托莫西汀
https://en.wikipedia.org/wiki/Atomoxetine
Azacitidine
阿扎胞苷
https://en.wikipedia.org/wiki/Azacitidine
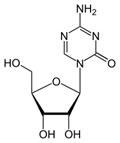
Azacitidine (INN; trade name Vidaza) is a
chemical analog of cytidine, a nucleoside in DNA and RNA. Azacitidine and its
deoxy derivative, decitabine (also known as 5-aza-2lso known as 5-aza-2′
Balsalazide Disodium Dihydrate
巴柳氮
二鈉二水合物https://en.wikipedia.org/wiki/Balsalazide
Balsalazide is an anti-inflammatory drug
used in the treatment of inflammatory bowel disease. It is sold under the brand
names Giazo, Colazal in the US and Colazide in the UK. It is also sold in
generic form in the US by several generic manufacturers.neric manufacturers.
Benazepril HCl
貝那普利
https://en.wikipedia.org/wiki/Benazepril
Benazepril, brand name Lotensin (Novartis),
is an ACE inhibitor used primarily in treatment of hypertension, congestive
heart failure, and heart attacks, and also in preventing the renal and retinal
complications of diabetes.cations of diabetes.
ACE inhibitors relax blood vessels, and
decrease blood volume, which lowers blood pressure and decreases oxygen demand
from the heart. They inhibit angiotensin-converting enzyme, which is part of the
renin–angiotensin–aldosterone system.
Benzonatate
苯甲酸鹽
https://en.wikipedia.org/wiki/Benzonatate
Benzonatate is a non-narcotic oral cough
suppressant, or antitussive, with effects that last from 6 to 8 hours. Since it
is not an opioid, benzonatate is not as prone to abuse like some other cough
medications such as codeine. Benzonatate was approved by the U.S. Food and Drug
Administration(FDA) in 1958.[1]
Bepotastine Besilate
苯磺酸贝他斯汀
https://en.wikipedia.org/wiki/Bepotastine
Bepotastine (Talion, Bepreve) is a 2nd
generation antihistamine.[1] It was approved in Japan for use in the treatment
of allergic rhinitis and urticaria/pruritus in July 2000 and January 2002,
respectively. It is currently marketed in the United States under the brand-name
Bepreve, by ISTA Pharmaceuticals.
Bimatoprost
比馬前列素
https://en.wikipedia.org/wiki/Bimatoprost
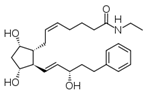
Bimatoprost (marketed in the US, Canada and
Europe by Allergan, under the trade name Lumigan) is a prostaglandin analog used
topically (as eye drops) to control the progression of glaucoma and in the
management of ocular hypertension. It reduces intraocular pressure (IOP) by
increasing the outflow of aqueous fluid from the eyes.[1] In December 2008, the
indication to lengthen eyelasheswas approved by the U.S. Food and Drug
Administration (FDA); the cosmetic formulation of bimatoprost is sold as Latisse
/ləˈtiːs/.[2]
Bisoprolol Fumarate
比索洛爾
富馬酸鹽
https://en.wikipedia.org/wiki/Bisoprolol

Bisoprolol is a drug belonging to the group
of beta-blockers, a class of medicines used primarily incardiovascular diseases.
More specifically, it is a selective type β1 adrenergic receptor blocker. The
U.S. Food and Drug Administration (FDA) approved an application by Duramed
Pharmaceutical for Zebeta Oral Tablets (bisoprolol fumarate) as a new molecular
entity on July 31, 1992.[4] It has since been approved by the FDA for
manufacture by Teva, Mylan, Sandoz, Aurobino, and Unichem.[5] It is on the World
Health Organization's List of Essential Medicines, the most important
medications needed in a basic health system.[6]
Bivalirudin
比伐盧定
https://en.wikipedia.org/wiki/Bivalirudin
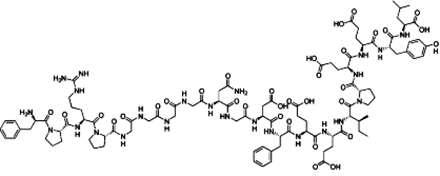
Bivalirudin (Angiomax or Angiox,
manufactured by The Medicines Company) is a specific and reversible direct
thrombin inhibitor (DTI).[1]
Chemically, it is a synthetic congener of
the naturally occurring drug hirudin (found in the saliva of the medicinal leech
Hirudo medicinalis).Hirudo medicinalis).
Bortezomib
硼替佐米
https://en.wikipedia.org/wiki/Bortezomib
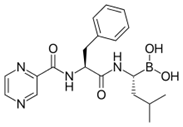
Bortezomib (BAN, INN and USAN. Originally
codenamed PS-341; marketed as Velcade byMillennium Pharmaceuticals; Neomib by
Getwell and Bortecad by Cadila Healthcare) is the first therapeutic proteasome
inhibitor to be tested in humans. Proteasomes are cellular complexes that break
down proteins. In some cancers, the proteins that normally kill cancer cells are
broken down too quickly. Bortezomib interrupts this process and lets those
proteins kill the cancer cells. It is approved in the U.S. for treating relapsed
multiple myeloma and mantle cell lymphoma.[1][2] In multiple myeloma, complete
clinical responses have been obtained in patients with otherwise refractory or
rapidly advancing disease.
Brinzolamide
布林唑胺
https://en.wikipedia.org/wiki/Brinzolamide
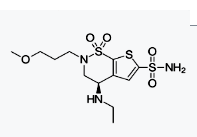
Brinzolamide (trade names Azopt, Alcon Laboratories, Befardin,[1] Fardi
Medicals,[2] ) is a carbonic anhydrase inhibitor used to lower intraocular
pressure in patients with open-angle glaucoma or ocular hypertension.
Chemistry[edit] Brinzolamide is a carbonic anhydrase inhibitor (specifically,
carbonic anhydrase II). Carbonic anhydrase is found primarily in erythrocytes
(but also in other tissues including the eye). It exists as a number of
isoenzymes, the most active of which is carbonic anhydrase II (CA-II).
Indications[edit] Use for the treatment of open-angle glaucoma and raised
intraocular pressure due to excess aqueous humor production.
Calcipotriol
卡泊三醇
Calcipotriol Monohydrate
https://en.wikipedia.org/wiki/Calcipotriol
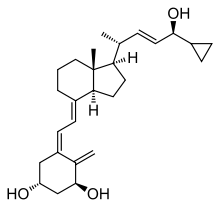
Calcipotriol (INN) or calcipotriene (USAN)
is a synthetic derivative of calcitriol, a form of vitamin D. It is used in the
treatment of psoriasis, marketed under the trade name "Dovonex" in the United
States, "Daivonex" outside of North America, and "Psorcutan" in Germany. This
medication is safe for long-term application in psoriatic skin conditions.
Calcitriol
骨化三醇
https://en.wikipedia.org/wiki/Calcitriol
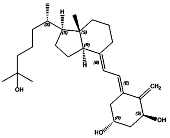
Calcitriol (INN), also called
1,25-dihydroxycholecalciferol or 1,25-dihydroxyvitamin D3, is the hormonally
active metabolite of vitamin D with three hydroxyl groups (abbreviated
1,25-(OH)2D3 or simply 1,25(OH)2D),[6] It was first identified by Michael F.
Holick in work published in 1971.[7] Calcitriol increases the level of calcium
(Ca2+) in the blood by increasing the uptake of calcium from the gut into the
blood, and possibly increasing the release of calcium into the blood from
bone.[8]
Calcium Acetate
醋酸鈣
https://en.wikipedia.org/wiki/Calcium_acetate
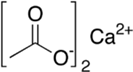
Calcium acetate is a chemical compound which
is a calcium salt of acetic acid. It has the formula Ca(C2H3O2)2. Its standard
name is calcium acetate, while calcium ethanoate is the systematic name. An
older name is acetate of lime. The anhydrous form is very hygroscopic; therefore
the monohydrate (Ca(CH3COO)2•H2O) is the common form. is the common form.
•
In kidney disease, blood levels of phosphate may
rise (called hyperphosphatemia) leading to bone problems. Calcium acetate binds
phosphate in the diet to lower blood phosphate levels.[citation needed]
Capsaicin
辣椒素
https://en.wikipedia.org/wiki/Capsaicin#Research_and_pharmaceutical_use
Capsaicin is used as an analgesic in topical
ointments, nasal sprays (Sinol-M), and dermal patches to relieve pain, typically
in concentrations between 0.025% and 0.1%.[38] It may be applied in cream form
for the temporary relief of minor aches and pains of muscles and joints
associated with arthritis, backache, strains and sprains, often in compounds
with other rubefacients.[38]
It is also used to reduce the symptoms of
peripheral neuropathy such as post-herpetic neuralgia caused by shingles.[38]
Capsaicin transdermal patch (Qutenza) for the management of this particular
therapeutic indication (pain due to post-herpetic neuralgia) was approved as a
therapeutic by the U.S. FDA,[39] but a subsequent application for Qutenza to be
used as an analgesic in HIV neuralgia was refused.[40]gia was refused.[40]
Although capsaicin creams have been used to
treat psoriasis for reduction of itching,[38][41][42] a review of six clinical
trials involving topical capsaicin for treatment of pruritus concluded there was
insufficient evidence of effect.[43]
Carbarsone
對脲苯基胂酸
https://en.wikipedia.org/wiki/Carbarsone

Carbarsone is an organoarsenic compound used
as an antiprotozoal drug for treatment of amebiasisand other
infections.[1][2][3] It was available for amebiasis in the United States as late
as 1991. Thereafter, it remained available as a turkey feed additive for
increasing weight gain and controllingblackhead disease.[4][5]khead disease.[4][5]
Caspofungin Acetate
乙酸卡泊芬淨
https://en.wikipedia.org/wiki/Caspofungin
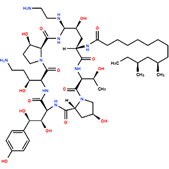
Caspofungin (INN)[1] (brand name Cancidas
worldwide) is a lipopeptide antifungal drug fromMerck & Co., Inc. discovered by
James Balkovec, Regina Black and Frances A. Bouffard.[2] It is a member of a new
class of antifungals termed the echinocandins. It works by inhibiting the
enzyme(1 →3)-β-D-glucan synthase and thereby disturbing the
integrity of the fungal cell wall. Caspofungin was the first inhibitor of fungal
(1→3)-β-D-glucan synthesis to be approved by the
United States Food and Drug Administration.[3] Caspofungin is administered
intravenously.
Cefaclor
頭孢克洛
https://en.wikipedia.org/wiki/Cefaclor
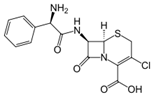
Cefaclor, developed by Eli Lilly under the
trade name Ceclor, is a second-generation cephalosporinantibiotic used to treat
some infections caused by bacteria such as pneumonia and infections of the ear,
lung, skin, throat, and urinary tract. It is also available from other
manufacturers as a generic.[1]
Cefaclor belongs to the family of
antibiotics known as the cephalosporins (cefalosporins). The cephalosporins are
broad-spectrum antibiotics that are used for the treatment of
septicaemia,pneumonia, meningitis, biliary tract infections, peritonitis, and
urinary tract infections. The pharmacology of the cephalosporins is similar to
that of the penicillins, excretion being principally renal. Cephalosporins
penetrate the cerebrospinal fluid poorly unless the meninges are
inflamed;cefotaxime is a more suitable cephalosporin than cefaclor for
infections of the central nervous system, e.g. meningitis. Cefaclor is active
against many bacteria, including both Gram-negative andGram-positive organisms.
Cephalexin
頭孢氨芐
https://en.wikipedia.org/wiki/Cefalexin
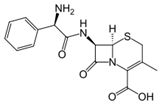
Cefalexin, also spelled cephalexin, is an
antibiotic that can treat a number of bacterial infections. It kills
gram-positive and some gram-negative bacteria by disrupting the growth of the
bacterial cell wall. Cefalexin is a beta-lactam antibiotic within the class of
first-generation cephalosporins.[3] It works similarly to other agents within
this class, including intravenous cefazolin, but can be taken by mouth.[4]e taken by mouth.[4]
Chlormezanone
氯苯甲酮
https://en.wikipedia.org/wiki/Chlormezanone
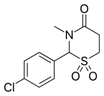
Chlormezanone (marketed under the brandname
Trancopal or Fenaprim) is a drug used as ananxiolytic and a muscle relaxant.d a muscle relaxant.
Chlorzoxazone
氯唑沙宗
https://en.wikipedia.org/wiki/Chlorzoxazone
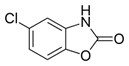
Chlorzoxazone (INN) is a centrally acting
muscle relaxant used to treat muscle spasm and the resulting pain or discomfort.
It acts on the spinal cord by depressing reflexes. It is sold under the trade
names "Lorzone", Paraflex and Muscol and in combination form as Parafon Forte, a
combination of chlorzoxazone and acetaminophen (paracetamol). Possible side
effects includedizziness, lightheadedness, malaise, nausea, vomiting, and liver
dysfunction. Used with acetaminophen it has added risk of hepatoxicity,[medical
citation needed] which is why the combination is not recommended. It can also be
administered for acute pain in general and for tension headache (muscle
contraction headache).ntraction headache).
Cilastatin Sodium
西司他汀鈉
https://en.wikipedia.org/wiki/Cilastatin
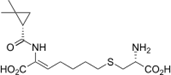
Cilastatin is a chemical compound which
inhibits the human enzyme dehydropeptidase.[1]dehydropeptidase.[1]
Clofibrate
氯貝特
https://en.wikipedia.org/wiki/Clofibrate
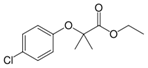
Clofibrate (tradename Atromid-S) is an
organic compound. It is marketed as a fibrate. It is a lipid-lowering agent used
for controlling the high cholesterol and triacylglyceride level in the blood. It
increases lipoprotein lipase activity to promote the conversion of VLDL to LDL,
and hence reduce the level of VLDL. It can increase the level of HDL as well.
Clonidine HCl
可樂定
https://en.wikipedia.org/wiki/Clonidine
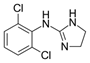
Clonidine (trade names Catapres, Kapvay,
Nexiclon, Clophelin, and others) is a medication used to treat high blood
pressure, attention deficit hyperactivity disorder, anxiety disorders,
withdrawal (from either alcohol, opioids, or smoking), migraine, menopausal
flushing, diarrhea, and certain pain conditions.[4] It is classified as a
centrally acting α2 adrenergic agonist and imidazoline receptoragonist that has
been in clinical use for over 40 years.[5]
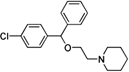
Cloperastine HCL
氯帕他汀
https://en.wikipedia.org/wiki/Cloperastine
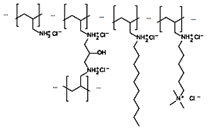
Colesevelam Hydrochloride
考來維侖
https://en.wikipedia.org/wiki/Colesevelam
The bile acid sequestrants are a group of
resins used to bind certain components of bile in the gastrointestinal tract.
They disrupt theenterohepatic circulation of bile acids by combining with bile
constituents and preventing their reabsorption from the gut. In general, they
are classified as hypolipidemic agents, although they may be used for purposes
other than lowering cholesterol. They are used in the treatment ofchronic
diarrhea due to bile acid malabsorption.
Bile acid sequestrants are polymeric
compounds that serve as ion-exchange resins. Bile acid sequestrants exchange
anions such as chloride ions for bile acids. By doing so, they bind bile acids
and sequester them from the enterohepatic circulation. The liver then produces
more bile acids to replace those that have been lost. Because the body uses
cholesterol to make bile acids, this reduces the amount of LDL cholesterol
circulating in the blood.[1] circulating in the blood.[1]
Bile acid sequestrants are large polymeric
structures, and they are not significantly absorbed from the gut into the
bloodstream. Thus, bile acid sequestrants, along with any bile acids bound to
the drug, are excreted via the feces after passage through the gastrointestinal
tract.[2]
Colestipol Hydrochloride
考來替泊
https://en.wikipedia.org/wiki/Colestipol
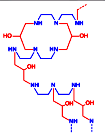
Colestipol (trade names Colestid,
Cholestabyl) is a bile acid sequestrant used to lower bloodcholesterol,
specifically low-density lipoprotein (LDL).[1][2] It is also used to reduce
stool volume and frequency, and in the treatment of chronic diarrhea.[3]
Like cholestyramine, colestipol works in the
gut by trapping bile acids and preventing them from being reabsorbed. This leads
to decreased enterohepatic recirculation of bile acids, increased synthesis of
new bile acids by the liver from cholesterol, decreased liver cholesterol,
increased LDL receptor expression, and decreasing LDL in blood.[4]
Crotamiton
克羅他米通
https://en.wikipedia.org/wiki/Crotamiton
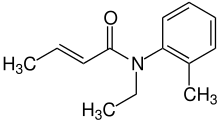
Crotamiton is a drug that is used both as a
scabicidal (for treating scabies) and as a general antipruritic (anti-itching
drug). It is a prescription lotion based medicine that is applied to the whole
body to get rid of the scabies parasite that burrows under the skin and causes
itching.
Pharmacology[edit]
The mechanism of action of crotamiton is
unknown, however it is toxic to the scabies mite.[2]
Cyclophosphamide
環磷酰胺
https://en.wikipedia.org/wiki/Cyclophosphamide
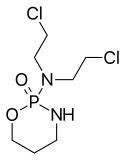
Cyclophosphamide (INN), also known as
cytophosphane,[3] is a medication mainly used inchemotherapy. It is an
alkylating agent of the nitrogen mustard type[4] (specifically, the
oxazaphosphorine group[5]).
An alkylating agent adds an alkyl group to
DNA. It attaches the alkyl group to the guanine base of DNA, at the number 7
nitrogen atom of the imidazole ring. This interferes with DNA replication by
forming intrastrand and interstrand DNA crosslinks.ntrastrand and interstrand DNA crosslinks.
Cyclophosphamide is used to treat cancers,
autoimmune disorders and AL amyloidosis. As aprodrug, it is converted by liver
cytochrome P450 (CYP) enzymes to form the metabolite 4-hydroxycyclophosphamide
that has chemotherapeutic activity.[6]
Dantrolene Sodium
丹曲林鈉
https://en.wikipedia.org/wiki/Dantrolene
Dantrolene sodium is a postsynaptic muscle
relaxant that lessens excitation-contraction coupling inmuscle cells. It
achieves this by inhibiting Ca2+ ions release from sarcoplasmic reticulum stores
by antagonizing ryanodine receptors.[1] It is the primary drug used for the
treatment and prevention ofmalignant hyperthermia, a rare, life-threatening
disorder triggered by general anesthesia. It is also used in the management of
neuroleptic malignant syndrome, muscle spasticity (e.g. after strokes,
inparaplegia, cerebral palsy, or patients with multiple sclerosis), and
2,4-dinitrophenol poisoning.[2] 2,4-dinitrophenol poisoning.[2]
It is marketed by JHP Pharmaceuticals LLC as
Dantrium (in North America) and by Norgine BV as Dantrium, Dantamacrin, or
Dantrolen (in Europe). A hospital is recommended to keep a minimum stock of 36
dantrolene vials (720 mg) sufficient for a 70-kg person.[3] As of 2015 the cost
for a typical course of medication in the United States is 100 to 200 USD.[4]
Daptomycin
達托黴素
https://en.wikipedia.org/wiki/Daptomycin
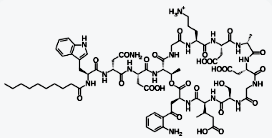
Daptomycin is a lipopeptide antibiotic used
in the treatment of systemic and life-threatening infections caused by
Gram-positive organisms. It is a naturally occurring compound found in the soil
saprotroph Streptomyces roseosporus. Its distinct mechanism of action makes it
useful in treating infections caused by multiple drug-resistant bacteria. It is
marketed in the United States under the trade name Cubicin by Cubist
Pharmaceuticals.
Mechanism of action[edit]
Daptomycin has a distinct mechanism of
action, disrupting multiple aspects of bacterial cell membrane function. It
inserts into the cell membrane in a phosphatidylglycerol-dependent fashion,
where it then aggregates. The aggregation of daptomycin alters the curvature of
the membrane, which creates holes that leak ions. This causes rapid
depolarization, resulting in a loss of membrane potential leading to inhibition
of protein, DNA, and RNA synthesis, which results in bacterial cell death.[4]
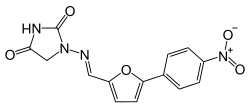
Decitabine
地西他濱
https://en.wikipedia.org/wiki/Decitabine
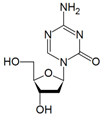
Decitabine is a hypomethylating agent.[3][4]
It hypomethylates DNA by inhibiting DNA methyltransferase.
It functions in a similar manner to
azacitidine, although decitabine can only be incorporated into DNA strands while
azacitidine can be incorporated into both DNA and RNA chains.
Clinical uses[edit]
Decitabine is indicated for the treatment of
myelodysplastic syndromes (MDS) including previously treated and untreated, de
novo and secondary MDS of all French-American-British subtypes (refractory
anemia, refractory anemia with ringed sideroblasts, refractory anemia with
excess blasts, refractory anemia with excess blasts in transformation, and
chronic myelomonocytic leukemia) and Intermediate-1, Intermediate-2, and
High-Risk International Prognostic Scoring System groups. In patients with renal
insufficiency, Batty and colleagues reported the first case series on the
feasibility of therapy with hypomethylating agents in patients with renal
insufficiency.[5]
It also has EU approval for acute myeloid
leukemia (AML).[2]
Desmopressin Acetate
去氨加壓素
https://en.wikipedia.org/wiki/Desmopressin
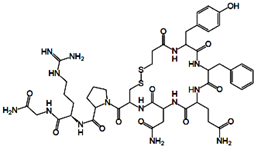
Desmopressin (trade names: DDAVP, others) is
a synthetic replacement for vasopressin, the hormone that reduces urine
production. It may be taken nasally, intravenously, or as an oral or sublingual
tablet. Physicians prescribe desmopressin most frequently for treatment of
diabetes insipidus, bedwetting, or nocturia, thrombocytopathy
It is on the WHO Model List of Essential
Medicines, the most important medications needed in a basic health system.[1]
Diclofenac Diethylamine
Diclofenac Epolamine
Diclofenac Potassium
Diclofenac Sodium
雙氯芬酸
https://en.wikipedia.org/wiki/Diclofenac
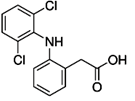
The primary mechanism responsible for its
anti-inflammatory, antipyretic, and analgesic action is thought to be inhibition
of prostaglandin synthesis by inhibition of cyclooxygenase (COX). It also
appears to exhibit bacteriostatic activity by inhibiting bacterial DNA
synthesis.[27]
Diclofenac (sold under a number of trade
names)[1] is a nonsteroidal anti-inflammatory drug (NSAID) taken or applied to
reduce inflammation and as an analgesic reducing pain in certain conditions. It
is supplied as or contained in medications under a variety of trade names.edications under a variety of trade names.
The name "diclofenac" derives from its
chemical name: 2-(2,6-dichloranilino) phenylacetic acid. Diclofenac was first
synthesized by Alfred Sallmann and Rudolf Pfister and introduced as Voltaren by
Ciba-Geigy (now Novartis) in 1973.[3]
In the United Kingdom, United States, India,
and Brazil diclofenac may be supplied as either the sodium or potassium salt; in
China, it is most often supplied as the sodium salt, while in some other
countries it is only available as the potassium salt. It is available as a
generic drug in a number of formulations, including diclofenac diethylamine,
which is applied topically.
Dimemorfan Phosphate
二甲啡烷
磷酸鹽
https://en.wikipedia.org/wiki/Dimemorfan
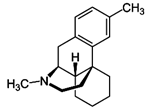
Dimemorfan (INN) (or dimemorphan) (brand
names Astomin, Dastosirr, Tusben), or dimemorfan phosphate (JAN), also known as
3,17-dimethylmorphinan, is an antitussive (cough suppressant) of the morphinan
family that is widely used in Japan and is also marketed in Spain and
Italy.[1][2][3][4] It was developed by Yamanouchi Pharmaceutical (now Astellas
Pharma) and introduced in Japan in 1975.[3] Dimemorfan is an analogue of
dextromethorphan(DXM) and its active metabolite dextrorphan (DXO), and similarly
to them, acts as a potent agonist of the σ1 receptor (Ki = 151
nM).[5][6]However, unlike DXM and DXO, it does not act significantly as an NMDA
receptor antagonist, and for this reason, lacks dissociativeeffects, thereby
having reduced side effects and abuse potential in comparison.[7][8] Similarly
to DXM and DXO, dimemorfan has only relatively low affinity for the σ2 receptor
(Ki = 4421 nM).[6]
Dinoprostone β-Cyclodextin
地諾前列酮
β-環糊精
Prostaglandin E2
前列腺素E2
https://en.wikipedia.org/wiki/Prostaglandin_E2
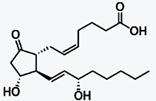
The naturally occurring prostaglandin E2
(PGE2 or PGE2) is known in medicine as dinoprostone. It has important effects in
labour (softening the cervix and causing uterine contraction) and also
stimulates osteoblasts to release factors that stimulate bone resorption by
osteoclasts. PGE2 is also the prostaglandin that ultimately induces fever.ostaglandin that ultimately induces fever.
PGE2 also suppresses T cell receptor
signaling and may play a role in resolution of inflammation.[1]
Cyclodextrin
https://en.wikipedia.org/wiki/Cyclodextrin
Cyclodextrins (sometimes called
cycloamyloses) are a family of compounds made up of sugar molecules bound
together in a ring (cyclic oligosaccharides).ether in a ring (cyclic oligosaccharides).
Cyclodextrins are produced from starch by
means of enzymatic conversion. They are used in food, pharmaceutical,[1] drug
delivery,[2] and chemical industries, as well as agriculture and environmental
engineering.
Cyclodextrins are composed of 5 or more
α-D-glucopyranoside units linked 1->4, as in amylose (a fragment of starch). The
5-membered macrocycle is not natural. Recently, the largest well-characterized
cyclodextrin contains 32 1,4-anhydroglucopyranoside units, while as a poorly
characterized mixture, at least 150-membered cyclic oligosaccharides are also
known. Typical cyclodextrins contain a number of glucose monomers ranging from
six to eight units in a ring, creating a cone shape:
Diphemanil Methylsulphate
敵草胺
https://en.wikipedia.org/wiki/Diphemanil_metilsulfate
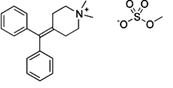
Diphemanil metilsulfate is an
antimuscarinic.
https://en.wikipedia.org/wiki/Muscarinic_antagonist
A muscarinic receptor antagonist (MRA) is a
type of anticholinergic agent that blocks the activity of the muscarinic
acetylcholine receptor. Acetylcholine (often abbreviated ACh) is a
neurotransmitter whose receptor is a protein found in synapses and other cell
membranes. Besides responding to their primary neurochemical, neurotransmitter
receptors can be sensitive to a variety of other molecules. Acetylcholine
receptors are classified into two groups based on this:
•
muscarinic, which respond to muscarine
•
nicotinic, which respond to nicotine
Most muscarinic receptor antagonists are
synthetic chemicals; however, the two most commonly used anticholinergics,
scopolamine and atropine, are belladonna alkaloids, and are naturally extracted.
Divalproex Sodium
雙丙戊酸鈉
https://en.wikipedia.org/wiki/Valproate
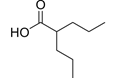
Valproate (VPA), and its valproic acid,
sodium valproate, and divalproex sodium forms, are medications primarily used to
treat epilepsy and bipolar disorder and to prevent migraine headaches.[2] It is
useful for the prevention of seizures in those with absence seizures, partial
seizures, and generalized seizures. It can be given intravenously or by mouth.
Long and short acting formulations exist.[2]
Mechanism of action[edit]
Although the mechanism of action of
valproate is not fully understood,[37] it has recently been shown to protect
against a seizure-induced reduction in phosphatidylinositol
(3,4,5)-trisphosphate (PIP3) as a potential therapeutic mechanism.[49] In
addition, its anticonvulsant effect has been attributed to the blockade of
voltage-dependent sodium channels and increased brain levels of
gamma-aminobutyric acid (GABA).[37] The GABAergic effect is also believed to
contribute towards the anti-manic properties of valproate.[37] In animals,
sodium valproate raises cerebral and cerebellar levels of the inhibitory
synaptic neurotransmitter, GABA, possibly by inhibiting GABA degradative
enzymes, such as GABA transaminase, succinate-semialdehyde dehydrogenase and by
inhibiting the re-uptake of GABA by neuronal cells.[37]
It also has histone deacetylase-inhibiting
effects. The inhibition of histone deacetylase, by promoting more
transcriptionally active chromatin structures, likely presents the epigenetic
mechanism for regulation of many of the neuroprotective effects attributed to
valproic acid. Intermediate molecules mediating these effects include VEGF,
BDNF, and GDNF.[50][51]ects attributed to
valproic acid. Intermediate molecules mediating these effects include VEGF,
Valproic acid has been found to be an
antagonist of the androgen and progesterone receptors, and hence as a
non-steroidal antiandrogen and antiprogestogen, at concentrations much lower
than therapeutic serum levels.[52] In addition, the drug has been identified as
a potent aromatase inhibitor, and suppresses estrogen concentrations.[53] These
actions are likely to be involved in the reproductive endocrine disturbances
seen with valproic acid treatment.[52][53]
DL-methylephedrine Hydrochloride
DL- 甲基麻黃鹼
鹽酸鹽
https://en.wikipedia.org/wiki/N-Methylephedrine
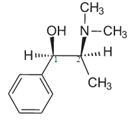
N-Methylephedrine is a derivative of
ephedrine. It has been isolated from Ephedra distachya.[2]
In organic chemistry, N-methylephedrine is
used as a resolving agent and as a precursor to chiral supporting electrolytes,
phase-transfer catalysts, and reducing agents.[3]https://en.wikipedia.org/wiki/Ephedrine
Ephedrine is a medication used to prevent
low blood pressure during spinal anesthesia.[1] It has also been used for
asthma, narcolepsy, and obesity but is not the preferred treatment.
Mechanism of action[edit]
Ephedrine, a sympathomimetic amine, acts on
part of the sympathetic nervous system (SNS). The principal mechanism of action
relies on its indirect stimulation of the adrenergic receptor system by
increasing the activity of norepinephrine at the postsynaptic α and β
receptors.[23] The presence of direct interactions with α receptors is unlikely,
but still controversial.[8][32][33] L-ephedrine, and particularly its
stereoisomer norpseudoephedrine (which is also present in Catha edulis) has
indirect sympathomimetic effects and due to its ability to cross the blood-brain
barrier, it is a CNS stimulant similar to amphetamines, but less pronounced, as
it releases noradrenaline and dopamine in the substantia nigra.[34]
The presence of an N-methyl group decreases
binding affinities at α receptors, compared with norephedrine. Ephedrine,
though, binds better than N-methylephedrine, which has an additional methyl
group at the nitrogen atom. Also the steric orientation of the hydroxyl group is
important for receptor binding and functional activity.[32]
Docetaxel
多西他賽
Docetaxel Trihydrate
https://en.wikipedia.org/wiki/Docetaxel
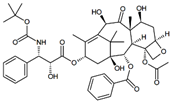
Docetaxel is a well-established anti-mitotic
chemotherapy medication that works by interfering with cell division. Docetaxel
is approved by the FDA for treatment of locally advanced or metastatic breast
cancer, head and neck cancer, gastric cancer, hormone-refractory prostate cancer
and non small-cell lung cancer.[1] Docetaxel can be used as a single agent or in
combination with other chemotherapeutic drugs as indicated depending on specific
cancer type and stage.[2]
Docetaxel is a member of the taxane drug
class, which also includes the chemotherapeutic medication paclitaxel.
Molecular target[edit]
Docetaxel binds to microtubules reversibly
with high affinity and has a maximum stoichiometry of 1 mole docetaxel per mole
tubulin in microtubules.[31] This binding stabilizes microtubules and prevents
depolymerisation from calcium ions, decreased temperature and dilution,
preferentially at the plus end of the microtubule.[31] Docetaxel has been found
to accumulate to higher concentration in ovarian adenocarcinoma cells than
kidney carcinoma cells, which may contribute to the more effective treatment of
ovarian cancer by docetaxel.[10][31] It has also been found to lead to the
phosphorylation of oncoprotein bcl-2, which is apoptosis-blocking in its
oncoprotein form.[10]
Modes of action[edit]
The cytotoxic activity of docetaxel is
exerted by promoting and stabilising microtubule assembly, while preventing
physiological microtubule depolymerisation/disassembly in the absence of
GTP.[10][16][32] This leads to a significant decrease in free tubulin, needed
for microtubule formation and results in inhibition of mitotic cell division
between metaphase and anaphase, preventing further cancer cell
progeny.[10][13][31]
Donepezil HCl
多奈哌齊
https://en.wikipedia.org/wiki/Donepezil
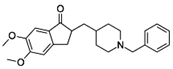
Donepezil, marketed under the trade name
Aricept, is a medication used in the palliative treatmentof Alzheimer's
disease.[1][2] Donepezil is used to improve cognition and behavior of people
with Alzheimer's, but does not slow the progression of or cure the disease.[3]Common side effects include loss of
appetite, gastrointestinal upset, diarrhea, difficulty sleeping, vomiting, or
muscle cramping.[4]
It was developed by Eisai and Pfizer and is
sold as a generic by multiple suppliers. Donepezil acts as a centrally acting
reversible acetylcholinesterase inhibitor.[5]
Doxercalciferol
骨化鈣醇
https://en.wikipedia.org/wiki/Doxercalciferol
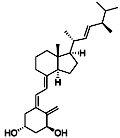
Doxercalciferol (trade name Hectorol) is
drug for secondary hyperparathyroidism and metabolic bone disease.[1] It is a
synthetic analog of ergocalciferol (vitamin D2). It suppresses
parathyroidsynthesis and secretion.[2]
Duloxetine hydrochloride
度洛西汀
鹽酸鹽
https://en.wikipedia.org/wiki/Duloxetine
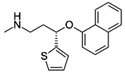
Duloxetine, sold under the brand name
Cymbalta among others,[1] is a serotonin–norepinephrine reuptake inhibitor
(SNRI). It is mostly prescribed for major depressive disorder, generalized
anxiety disorder, fibromyalgia and neuropathic pain.[2] (SNRI). It is mostly prescribed for major depressive disorder, generalized
anxiety disoDuloxetine failed to receive US approval for
stress urinary incontinence amid concerns over liver toxicity and suicidal
events; however, it was approved for this indication in the UK, where it is
recommended as an add-on medication in stress urinary incontinence instead of
surgery.[3] It was originally made by Eli Lilly.
Econazole Nitrate
https://en.wikipedia.org/wiki/Econazole
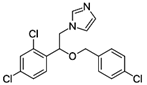
Econazole (commonly used as the nitrate
salt) is an antifungal medication of the imidazole class.[1]
Pharmaceutical derivatives[edit]
The substituted imidazole derivatives are
valuable in treatment of many systemic fungal infections.[17] Imidazoles belong
to the class of azole antifungals
https://en.wikipedia.org/wiki/Antifungal#Imidazole.2C_triazole.2C_and_thiazole_antifungals
Azole antifungal drugs (except for
abafungin) inhibit the enzyme lanosterol 14 α-demethylase; the enzyme necessary
to convert lanosterol to ergosterol. Depletion of ergosterol in fungal membrane
disrupts the structure and many functions of fungal membrane leading to
inhibition of fungal growth.[2]
Eflornithine Hydrochloride
地普崙胺
https://en.wikipedia.org/wiki/Eflornithine
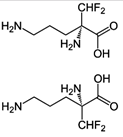
Eflornithine (α-difluoromethylornithine or
DFMO) is a drug found to be effective in the treatment of facial hirsutism[1]
(excessive hair growth) as well as in African trypanosomiasis (sleeping
sickness).[2] Eflornithine hydrochloride cream for topical application is meant
for women affected by facial hirsutism.
Description[edit]
Eflornithine is a "suicide inhibitor,"
irreversibly binding to Ornithine decarboxylase (ODC) and preventing the natural
substrate ornithine from accessing the active site (Figure 1). Within the active
site of ODC, eflornithine undergoes decarboxylation with aid of the cofactor
pyridoxal 5’-phosphate (PLP). Because of its additional difluoromethyl group in
comparison to ornithine, eflornithine is able to bind to a neighboring Cys-360
residue, permanently remaining fixated within the active site.
Entacapone
恩他卡朋
https://en.wikipedia.org/wiki/Entacapone
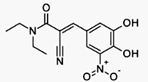
Entacapone (INN) is a medication commonly
used in combination with other medications for the treatment of Parkinson's
disease.[1] Entacapone together with levodopa and carbidopa allows levodopa to
have a longer effect in the brain and reduces Parkinson’s disease signs and
symptomsfor a greater length of time than levodopa and carbidopa therapy
alone.[1]
Entacapone is known as a selective and
reversible inhibitor of the enzyme catechol-O-methyltransferase (COMT).[1] When
taken together with levodopa (L-DOPA) and carbidopa, entacapone stops
catechol-O-methyltransferase from breaking down and metabolizing levodopa,
resulting in an overall increase of levodopa remaining in the brain and body.[1]Mechanism of action[edit]
Entacapone is a selective and reversible
inhibitor of catechol-O-methyltransferase (COMT).[1] COMT eliminates
biologically active catecholspresent in catecholamines (dopamine,
norepinephrine, and epinephrine) and their hydroxylated metabolites. When
administered with a decarboxylase inhibitor, COMT acts as the major metabolizing
enzyme for levodopa and metabolizes it to
3-methoxy-4-hydroxy-L-phenylalanine(3-OMD) in the brain and in the periphery.[1]
For the treatment of Parkinson’s disease,
entacapone is given as an adjunct to levodopa and an aromatic amino acid
decarboxylase inhibitor, carbidopa. Entacapone inhibits COMT and the metabolism
of levodopa, thus increasing plasma levels of levodopa and causing more constant
dopaminergic stimulation in order to reduce the signs and symptoms presented in
the disease.[1]
Entecavir monohydrate
恩替卡韋一水合物
https://en.wikipedia.org/wiki/Entecavir
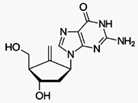
Entecavir (ETV), is an antiviral medication
used in the treatment of hepatitis B virus (HBV) infection. It is taken by
mouth. Entecavir is a reverse transcriptase inhibitor. It prevents the hepatitis
B virus from multiplying and reduces the amount of virus in the body.[1]
Entecavir is a nucleoside analog,[7] More
specifically, it is a deoxyguanosine analogue belonging to a class of
carbocyclic nucleosides, that inhibits reverse transcription, DNA replication
and transcription in the viral replication process.
Eperisone HCl
丙哌維酮
https://en.wikipedia.org/wiki/Eperisone
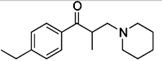
Eperisone (formulated as the eperisone
hydrochloride salt) is an antispasmodic drug.
Eperisone acts by relaxing both skeletal
muscles and vascular smooth muscles, and demonstrates a variety of effects such
as reduction of myotonia, improvement of circulation, and suppression of the
pain reflex. The drug inhibits the vicious circle of myotonia by decreasing
pain, ischaemia, and hypertonia in skeletal muscles, thus alleviating stiffness
and spasticity, and facilitating muscle movement[1]
Eperisone also improves dizziness and
tinnitus associated with cerebrovascular disorders or cervical spondylosis.
Ephedrine Hydrochloride
麻黃素
https://en.wikipedia.org/wiki/Ephedrine
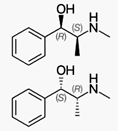
Ephedrine is a medication used to prevent
low blood pressure during spinal anesthesia.[1] It has also been used for
asthma, narcolepsy, and obesity but is not the preferred treatment. It can be
taken by mouth or by injection into a muscle, vein, or just under the skin.
Onset with intravenous use is fast, while injection into a muscle can take 20
minutes, and by mouth can take an hour for effect. When given by injection it
lasts about an hour and when taken by mouth it can last up to four hours.[1]or just under the skin.
Onset with intravenous use is fast, while injection into a muscle can take 20
minutes, and by mouth can take an hour for effect. When given by injection it
lasts about an hour and when taken by mouth it can last up to four hours.[1]
Ephedrine, a sympathomimetic amine, acts on
part of the sympathetic nervous system (SNS). The principal mechanism of action
relies on its indirect stimulation of the adrenergic receptor system by
increasing the activity of norepinephrine at the postsynaptic α and β
receptors.[23] The presence of direct interactions with α receptors is unlikely,
but still controversial.[8][32][33] L-ephedrine, and particularly its
stereoisomer norpseudoephedrine (which is also present in Catha edulis) has
indirect sympathomimetic effects and due to its ability to cross the The presence of an N-methyl group decreases
binding affinities at α receptors, compared with norephedrine. Ephedrine,
though, binds better than N-methylephedrine, which has an additional methyl
group at the nitrogen atom. Also the steric orientation of the hydroxyl group is
important for receptor binding and functional activity.[32]
Epoprostenol Sodium
依前列醇鈉
https://en.wikipedia.org/wiki/Prostacyclin
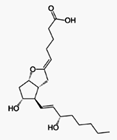
Prostacyclin (also called prostaglandin I2
or PGI2) is a prostaglandin member of the eicosanoid family of lipid molecules.
It inhibits platelet activation and is also an effective vasodilator.
Prostacyclin (also called prostaglandin I2
or PGI2) is a prostaglandin member of the eicosanoid family of lipid molecules.
It inhibits platelet activation and is also an effective vasodilator.
Ertapenem
厄他培南
https://en.wikipedia.org/wiki/Ertapenem
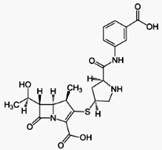
Ertapenem is a carbapenem antibiotic
marketed by Merck as Invanz. It is structurally very similar to meropenem in
that it possesses a 1-β-methyl group. Other members of the carbapenem group
(imipenem, doripenem, and meropenem) are broadly active antibacterials that are
used for infections caused by difficult to treat or multidrug-resistant bacteria
(such as ESBL expressing Klebsiella pneumonia). They have very short serum
half-lives and must be administered by intravenous infusion every 6 to 8 hours.
Ertapenem differs from other carbapenems in having a somewhat less broad
spectrum of activity (not against Pseudomonas aeruginosa), and in that its
extended serum half-life allows it to be administered once every 24 hours.[1]
Ethoxybenzamide
乙氧基苯甲酰胺
https://en.wikipedia.org/wiki/Ethenzamide
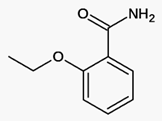
Ethenzamide, Systematic (IUPAC) name
2-ethoxybenzamide, is a ommon analgesic and anti-inflammatory drug that is used
for the relief of fever, headaches, and other minor aches and pains.[1][2] It is
an ingredient in numerous cold medications and many prescription analgesics.
Ethyl Icosapentate
二十碳五烯酸乙酯
ttps://en.wikipedia.org/wiki/Ethyl_eicosapentaenoic_acid
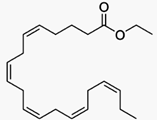
Ethyl eicosapentaenoic acid (E-EPA,
icosapent ethyl) is a
derivative of the
omega-3 fatty acid
eicosapentaenoic acid
(EPA) that is used in combination with changes in diet to lower
triglyceridelevels in adults with severe (≥ 500 mg/dL)
hypertriglyceridemia.
This was the second class of fish oil-based drug to be approved for use as a
drug and was approved by the FDA in 2012. These fish oil drugs are similar to
fish oil dietary supplements but the ingredients are better controlled and have
been tested in clinical trials. he second class of fish oil-based drug to be approved for use as a
drug and was approved by the FDA in 2012. These fish oil drugs are similar to
fish oil dietary supplements but the ingredients are better controlled and have
been tested in clinical trials.
The company that developed this drug,
Amarin
Corporation, challenged the FDA's ability to limit its ability to market the
drug for off-label use
and won its case on appeal in 2012, changing the way the FDA regulates
pharmaceutical marketing.
The active metabolite of E-EPA, like
other omega-3 fatty acid based drugs, appears to reduce production of
triglycerides in the liver, and to enhance clearance of triglycerides from
circulating very low-density lipoprotein (VLDL) particles; the way it does that
is not clear, but potential mechanisms include increased breakdown of fatty
acids; inhibition of diglyceride acyltransferase which is involved in
biosynthesis of triglycerides in the liver; and increased activity of
lipoprotein lipase in blood.[1][3]
15 Health Benefits Of Omega-3 Fatty
Acids, According To Science (+15 Best Omega-3 Foods)
https://www.jenreviews.com/omega-3/

Omega-3 is important for quite a few reasons, some of which
being;
Regulation of blood clotting
Boosting artery wall relaxation and contraction
Strengthening of cell membranes
Normalizing the speed at which your heart beats
Brain development and growth
And that’s just the beginning. If you're not reaching for a plate of salmon just
yet, you will be after this list!
1. Omega-3 Fatty Acids Helps Fight Anxiety
2. Omega-3 Fatty Acids Improve Eye Health
3. Omega-3 Fatty Acids Can Help Fight Infertility
4. Omega-3 Fatty Acids Can Be Used To Treat Skin Issues
5. Omega-3 Fatty Acids Improve Cardiovascular health
6. Omega-3 Fatty Acids Can Ease Menstrual Pain
7. Omega-3 Fatty Acids Reduces Fatty Liver
8. Omega-3 Fatty Acids Prevents And Reverses Insulin Resistance
9. Omega-3 Fatty Acids Lowers Cholesterol
10. Omega-3 Fatty Acids Protects And Improves Brain Health During Pregnancy
11. Omega-3 Fatty Acids Counters Inflammation
12. Omega-3 Fatty Acids Fights Autoimmune Disease
13. Omega-3 Fatty Acids Improves Sleep Patterns
14. Omega-3 Fatty Acids Help With Symptoms Of ADHD
15. Omega-3 Fatty Acids Can Be Beneficial To Persons Living With Asthma
15 Best Omega-3 Fatty Acids Rich Foods :
1. Salmon
2. Chia seeds
3. Walnuts
4. Egg Yolks
5. Spinach
6. Fish Oil
7. Soybeans
8. Mackerel
9. Flaxseeds
10. Oysters
11. Anchovies
12. Tofu
13. Lentils
14. Mustard seeds
15. Kidney Beans
Everolimus
依維莫司
Everolimus B20
https://en.wikipedia.org/wiki/Everolimus
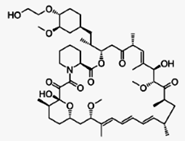
Everolimus (INN) (/ˌɛvəˈroʊləməs/) (earlier
code name RAD001) is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works
similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).ass="MsoNormal">
Everolimus (INN) (/ˌɛvəˈroʊləməs/) (earlier IIt is currently used as an immunosuppressant
to prevent rejection of organ transplants and treatment of renal cell cancer and
other tumours. Much research has also been conducted on everolimus and other
mTOR inhibitors as targeted therapy for use in a number of cancers.
Exemestane
依西美坦
https://en.wikipedia.org/wiki/Exemestane
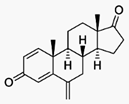
Exemestane (trade name Aromasin) is a drug
used to treat breast cancer. It is a member of the class of drugs known as
aromatase inhibitors. Some breast cancers require estrogen to grow. Those
cancers have estrogen receptors (ERs), and are called ER-positive. They may also
be called estrogen-responsive, hormonally-responsive, or
hormone-receptor-positive. Aromatase is an enzyme that synthesizes estrogen.
Aromatase inhibitors block the synthesis of estrogen. This lowers the estrogen
level, and slows the growth of cancers.
Famotidine
法莫替丁
https://en.wikipedia.org/wiki/Famotidine
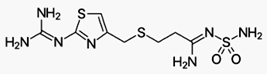
Famotidine, sold under the trade name Pepcid
among others is a histamine H2 receptor antagonist that inhibits stomach acid
production. It is commonly used in the treatment of peptic ulcer disease and
gastroesophageal reflux disease.
Unlike cimetidine, the first H2 antagonist,
famotidine has no effect on the cytochrome P450enzyme system, and does not
appear to interact with other drugs.[2]
It was discovered in 1979.[3]
https://en.wikipedia.org/wiki/H2_antagonist
H2 antagonists, also called H2 blockers, are
a class of medications that block the action of histamine at the histamine H2
receptors of the parietal cells in the stomach. This decreases the production of
stomach acid. H2 antagonists can be used in the treatment of dyspepsia, but have
been surpassed by the more effective[1] proton pump inhibitors. They are also
used to treat peptic ulcer disease and gastroesophageal reflux disease
Felodipine
非洛地平
https://en.wikipedia.org/wiki/Felodipine
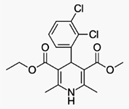
Felodipine is a calcium channel blocker
(calcium antagonist), a drug used to control hypertension(high blood pressure).
Felodipine is a calcium channel blocker.
https://en.wikipedia.org/wiki/Calcium_channel_blocker
Felodipine has additionally been found to
act as an antagonist of the mineralocorticoid receptor, or as an
antimineralocorticoid.[4]
Flavoxate Hydrochloride
氟沙星
https://en.wikipedia.org/wiki/Flavoxate
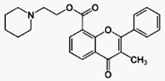
Flavoxate is an anticholinergic with
antimuscarinic effects. Its muscle relaxant properties may be due to a direct
action on the smooth muscle rather than by antagonizing muscarinic receptors.
Flavoxate is an anticholinergic with
antimuscarinic effects. Its muscle relaxant properties may be due to a direct
action on the smooth muscle rather than by antagonizing muscarinic receptors.
Flavoxate is used to treat urinary bladder
spasms. It is available under the trade name Urispas (Paladin),Genurin (by
Recordati, Italy) in Flavoxate is indicated for symptomatic
relief of interstitial cystitis, dysuria, urgency, nocturia, suprapubic pain,
frequency and incontinence as may occur in cystitis, prostatitis, urethritis,
urethrocystitis/urethrotrigonitis.
Fluconazole
氟康唑
https://en.wikipedia.org/wiki/Fluconazole
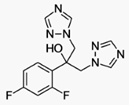
Fluconazole is an antifungal medication that
is given either by mouth or intravenously. It is used to treat a variety of
fungal infections, especially Candida infections of the vagina (yeast
infections), mouth, throat, and bloodstream. It is also used to prevent
infections in people with weak immune systems, including those with neutropenia
due to cancer chemotherapy, transplant patients, and premature babies.
In those who are pregnant it may increase
the risk of miscarriage.[1]
Mechanism of action[edit]
Like other imidazole- and triazole-class
antifungals, fluconazole inhibits the fungal cytochrome P450 enzyme
14α-demethylase. Mammalian demethylase activity is much less sensitive to
fluconazole than fungal demethylase. This inhibition prevents the conversion of
lanosterol to ergosterol, an essential component of the fungal cytoplasmic
membrane, and subsequent accumulation of 14α-methyl sterols.[16] Fluconazole is
primarily fungistatic; however, it may be fungicidal against certain organisms
in a dose-dependent manner, specifically Cryptococcus.[25]
It is interesting to note, when fluconazole
was in development at Pfizer, it was decided early in the process to avoid
producing any chiral centers in the drug so subsequent synthesis and
purification would not encounter difficulties with enantiomer separation and
associated variations in biological effect.[citation needed] A number of related
compounds were found to be extremely potent teratogens, and were subsequently
discarded.[citation needed]
Fludiazepam
氟脲重
https://en.wikipedia.org/wiki/Fludiazepam
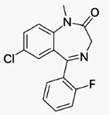
Fludiazepam, marketed under the brand name
Erispan ( エリスパン)[1][2] is a potent benzodiazepine and
2ʹ-fluoro derivative of diazepam,[3] originally developed by Hoffman-La Roche in
the 1960s.[4] It is marketed in Japan and Taiwan.[citation needed] It exerts its
pharmacological properties via enhancement of GABAergic inhibition.[5]
Fludiazepam has 4 times more binding affinity for benzodiazepine receptors than
diazepam.[6] It possesses anxiolytic,[7][8][9] anticonvulsant, sedative,
hypnotic and skeletal muscle relaxant properties.[10]
As with all benzodiazepines, fludiazepam is
used recreationally.[11]
Flumazenil
氟馬西尼
https://en.wikipedia.org/wiki/Flumazenil
Flumazenil (also known as flumazepil, code
name Ro 15-1788) is a selective benzodiazepine receptor antagonist[1] primarily
available by injection only. It has antagonistic and antidote properties to
therapeutically used benzodiazapenes, through competitive inhibition.lso known as flumazepil, code
name Ro 15-1788) is a selective benzodiazepine receptor antagonist[It was first introduced in 1987 by
Hoffmann-La Roche under the trade name Anexate, but only approved by the FDA on
December 20, 1991. Flumazenil went off patent in 2008 so at present generic
formulations of this drug are available. Intravenous flumazenil is primarily
used to treat benzodiazepine overdoses and to help reverse anesthesia.
Administration of flumazenil by sublingual lozenge and topical cream has also
been tested.[2][3]
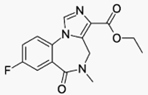
Flupentixol Dihydrochloride
氟達醇
二鹽酸鹽
https://en.wikipedia.org/wiki/Flupentixol
Flupentixol (INN), also known as
flupenthixol (former BAN), marketed under brand names such as Depixol and
Fluanxol is a typical antipsychotic drug of the thioxanthene class. It was
introduced in 1965 by Lundbeck. In addition to single drug preparations, it is
also available as flupentixol/melitracen—a combination product containing both
melitracen (a tricyclic antidepressant) and flupentixol. Flupentixol is not
approved for use in the United States. It is, however, approved for use in the
UK,[4] Australia,[5] Canada, Russian Federation,[6] South Africa, New Zealand,
Philippinesand various other countries.
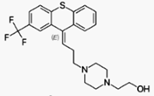
Fondaparinux Sodium
磺達肝素鈉
https://en.wikipedia.org/wiki/Fondaparinux
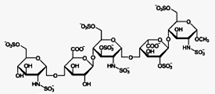
Fondaparinux (trade name Arixtra) is an
anticoagulant medication chemically related to low molecular weight heparins. It
is marketed by GlaxoSmithKline. A generic version developed by Alchemia is
marketed within the US by Dr. Reddy's Laboratories.
Fondaparinux is a synthetic pentasaccharide
factor Xa inhibitor. Apart from the O-methyl group at the reducing end of the
molecule, the identity and sequence of the five monomeric sugar units contained
in fondaparinux is identical to a sequence of five monomeric sugar units that
can be isolated after either chemical or enzymatic cleavage of the polymeric
glycosaminoglycans heparinand heparin sulfate (HS). Within heparin and heparin
sulfate this monomeric sequence is thought to form the high-affinity binding
site for the anti-coagulant factor antithrombin III (ATIII). Binding of
heparin/HS to ATIII has been shown to increase the anti-coagulant activity of
antithrombin III 1000 fold. In contrast to heparin, fondaparinux does not
inhibit thrombin.
Fulvestrant 氟維司群
https://en.wikipedia.org/wiki/Fulvestrant
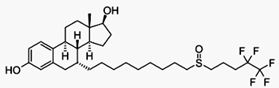
Fulvestrant (trade name Faslodex, by AstraZeneca) is a drug treatment of hormone
receptor-positive metastatic breast cancer in postmenopausal women with disease
progression following anti-estrogen therapy. It is a complete estrogen receptor
antagonist with no agonist effects, which in addition, accelerates the
proteasomal degradation of the estrogen receptor.[1] The drug has poor oral
bioavailability, and is administered monthly via intramuscular injection.[2]
Fursultiamine Hydrochloride (Fulfultiamine 鹽酸鹽)
Fursultiamine powder
Thiamine Tetrahydrofurfuryl Disulfide
維生素B1衍生物
https://en.wikipedia.org/wiki/Fursultiamine
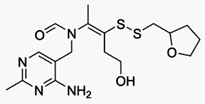
Fursultiamine (INN; Adventan, Alinamin-F, Benlipoid, Bevitol Lipophil, Judolor),
also known as thiamine tetrahydrofurfuryl disulfide (TTFD), is a disulfide
derivative of thiamine, or an allithiamine.[1] It was synthesized in Japan in
the 1960s for the purpose of developing forms of thiamine with improved
lipophilicity for treating vitamin B1 deficiency (i.e., beriberi),[1][2] and was
subsequently commercialized not only in Japan but also in Spain, Austria,
Germany, and the United States.[3] As a vitamin, it is available
over-the-counter as well.[4]
In addition to its clinical indication of avitaminosis, fursultiamine has been
studied in clinical trials for Alzheimer's disease and autistic spectrum
disorders with positive but modest benefits.[5][6] It has also been investigated
in improving energy metabolism during exercise and reducing exercise-induced
fatigue with conflicting results.[4][7][8][9]
Gadodiamide Hydrate
甘二酰胺水合物
https://en.wikipedia.org/wiki/Gadodiamide
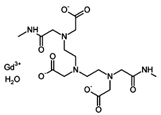
Gadodiamide is a gadolinium-based MRI
contrast agent, used in MR imagingprocedures to assist in the visualization of
blood vessels. It is commonly marketed under the trade name Omniscan.
A 2015 study found trace amounts of
Gadolinium deposited in the brain tissue of patients that had received
Gadodiamide.[1][2]
Gadodiamide is a contrast medium for cranial
and spinal magnetic resonance imaging(MRI) and for general MRI of the body after
intravenous administration. The product provides contrast enhancement and
facilitates visualisation of abnormal structures or lesions in various parts of
the body including the central nervous system (CNS). It does not cross an intact
blood brain barrier but might give enhancement in pathological conditions.
Gadopentetate Dimeglumine
戊二酸二甲葡胺https://en.wikipedia.org/wiki/Gadopentetic_acid
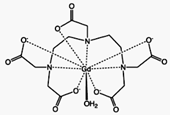
Gadopentetic acid is one of the trade names
for a gadolinium-based MRI contrast agent, usually administered as a salt of a
complex of gadolinium with DTPA (diethylenetriaminepentacetate) with the
chemical formula A2[Gd(DTPA)(H2O)]; when cation A is the protonated form of the
amino sugarmeglumine the salt goes under the name "gadopentetate dimeglumine".
It was described in 1981 and introduced as the first MRI contrast agent in 1987.
It is used to assist imaging of blood vessels and of inflamed or diseased tissue
where the blood vessels become "leaky". It is often used when viewing
intracranial lesions with abnormal vascularity or abnormalities in the
blood–brain barrier. It is usually injected intravenously. Gd-DTPA is classed as
an acyclic, ionic gadolinium contrast medium. Its paramagnetic property reduces
the T1 relaxation time (and to some extent the T2 and T2* relaxation times) in
NMR, which is the source of its clinical utility.
Gadoterate Meglumine
釓特酸葡甲胺
https://en.wikipedia.org/wiki/Gadoteric_acid
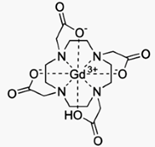
Gadoteric acid (trade names Artirem, Dotarem
or Dotagita) is a macrocycle-structured gadolinium-based MRI contrast agent. It
consists of the organic acid DOTA as a chelating agent, and gadolinium (Gd3+),
and is used in form of the meglumine salt (Gadoterate).[1] The drug is approved
and used in a number of countries worldwide.[2] It is used to assist imaging of
blood vessels and of inflamed or diseased tissue where the blood vessels become
'leaky'. It is often used when viewing intracranial lesions with abnormal
vascularity or abnormalities in the blood–brain barrier. Gadoteric acid is used
for MRI imaging of the brain, spine, and associated tissues for adult and
pediatric (2 year of age or older) patients.[3] The meglumine salt it takes the
form of crosses the blood brain barrier of tissue with abnormal vasculature,
highlighting the affected area with MRI. Gadoterate does not cross the intact
blood-brain barrier, so it does not affect or enhance normal brain tissue in
imaging [3].
Galantamine HBr
加蘭他敏
https://en.wikipedia.org/wiki/Galantamine
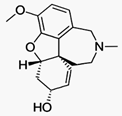
Galantamine (Nivalin, Razadyne, Razadyne ER,
Reminyl, Lycoremine) is used for the treatment of mild to moderate Alzheimer's
disease and various other memory impairments, in particular those of vascular
origin. It is an alkaloid that is obtained synthetically or from the bulbs and
flowers of Galanthus caucasicus (Caucasian snowdrop, Voronov's snowdrop),
Galanthus woronowii(Amaryllidaceae) and related genera like Narcissus
(daffodil), Leucojum aestivum (snowflake), and Lycoris including Lycoris radiata
(red spider lily).[1]ious other memory impairments, in particular those of vascular
origin. It is an alkaloid that is Studies of usage in modern medicine began in
the Soviet Union in the 1950s. The active ingredient was extracted, identified,
and studied, in particular in relation to its acetylcholinesterase
(AChE)-inhibiting properties. The bulk of the work was carried out by Soviet
pharmacologists M. D. Mashkovsky and R. P. Kruglikova–Lvova, beginning in
1951.[2] The work of Mashkovsky and Kruglikova-Lvova was the first published
work that demonstrated the AChE-inhibiting properties of galantamine.[3]
Gemcitabine HCl
吉西他濱
https://en.wikipedia.org/wiki/Gemcitabine
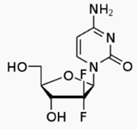
Gemcitabine (pronunciation: jem-SITE-a-been)
is a nucleoside analog used in chemotherapy. It is marketed as Gemzar by Eli
Lilly and Company.://en.wikipedia.org/wiki/Gemcitabine
Gemcitabine (pronunciation: jem-SITE-a-been)
is a nucleoside analog used in chemotherapy. It is marketed as Gemzar by Eli
Lilly and Company.
It is on the WHO Model List of Essential
As with fluorouracil and other analogues of
pyrimidines, the triphosphate analogue of gemcitabine replaces one of the
building blocks of nucleic acids, in this case cytidine, during DNA replication.
The process arrests tumor growth, as only one additional nucleoside can be
attached to the "faulty" nucleoside, resulting in apoptosis.
Another target of gemcitabine is the enzyme
ribonucleotide reductase (RNR). The diphosphate analogue binds to RNR active
site and inactivates the enzyme irreversibly. Once RNR is inhibited, the cell
cannot produce the deoxyribonucleotides required for DNA replication and repair,
and cell apoptosis is induced.[5]
Gimeracil
吉莫斯特
https://en.wikipedia.org/wiki/Tegafur/gimeracil/oteracil
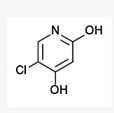
Mechanism of action[edit]
Tegafur is the actual chemotherapeutic
agent. It is a prodrug of the active substance fluorouracil (5-FU).
Gimeracil inhibits the degradation of
fluorouracil by reversibly blocking a dehydrogenase enzyme. This results in
higher 5-FU levels and a prolonged half-life of the substance.
Oteracil mainly stays in the gut because of
its low permeability, where it reduces the production of 5-FU by blocking the
enzyme orotate phosphoribosyltransferase. Lower 5-FU levels in the gut result in
a lower gastrointestinal toxicity.[8]
Glyceryl Guaiacolate
甘油癒創木酚
Guaifenesin
https://en.wikipedia.org/wiki/Guaifenesin
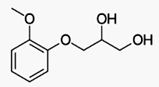
Guaifenesin INN /ɡwaɪˈfɛnᵻsɪn/ or
guaiphenesin (former BAN), also glyceryl guaiacolate,[2] is an expectorant drug
sold over the counter and usually taken orally to assist the bringing up
(expectoration) of phlegm from the airways in acute respiratory tract
infections.
Mechanism of action[edit]
Guaifenesin is thought to act as an
expectorant by increasing the volume and reducing the viscosity of secretions in
the trachea and bronchi. It has been said to aid in the flow of respiratory
tract secretions, allowing ciliary movement to carry the loosened secretions
upward toward the pharynx.[12] Thus, it may increase the efficiency of the cough
reflex and facilitate removal of the secretions.
Guaifenesin has muscle relaxant and
anticonvulsant properties and may be acting as an NMDA receptor antagonist.[13]
Granisetron Base
格拉司瓊
Granisetron Hydrochloride
https://en.wikipedia.org/wiki/Granisetron
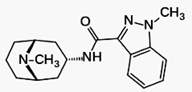
Granisetron is a serotonin 5-HT3 receptor
antagonist used as an antiemetic to treat nausea and vomiting following
chemotherapy
Granisetron is a serotonin 5-HT3 receptor
antagonist used as an antiemetic to treat nausea and vomiting following
chemotherapy. Its main effect is to reduce the activity of the vagus nerve,
which is a nerve that activates the vomiting center in the medulla oblongata. It
does not have much effect on vomiting due to motion sickness. This drug does not
have any effect on dopamine receptors or muscarinic receptors.
Hydroxychloroquine Sulfate
羥基氯喹
硫酸鹽https://en.wikipedia.org/wiki/Hydroxychloroquine
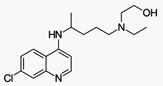
Hydroxychloroquine (HCQ), sold under the
trade names Plaquenil among others, is an antimalarial medication.
It is also
used to reduce inflammation in the treatment of rheumatoid arthritis(see
disease-modifying antirheumatic drugs) and lupus. Hydroxychloroquine differs
from chloroquineby the presence of a hydroxyl group at the end of the side
chain: the N-ethyl substituent is beta-hydroxylated. It is available for
administration by mouth as hydroxychloroquine sulfate. an antimalarial medication.
It is also
used to reduce inflammation in the treatment of rheumatoid arthritis(see
disease-modifying antirheumatic drugs) and lupus. Hydroxychloroquine differs
from chloroquineby the presence of a hydroxyl group at the end of the side
chain: the N-ethyl substituent is beta-hydroxylated.
It is available for
As with other quinoline antimalarial drugs,
the mechanism of action of quinine has not been fully resolved.
The most
accepted model is based on hydrochloroquinine, and involves the inhibition of
hemozoin biocrystallization, which facilitates the aggregation of cytotoxic
heme. Free cytotoxic heme accumulates in the parasites, causing their deaths.[c]
Imatinib Mesylate 伊馬替尼
甲磺酸鹽
https://en.wikipedia.org/wiki/Imatinib
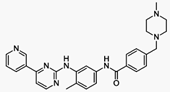
Imatinib, sold under the brand names
Gleevec and Glivec, used in the treatment of multiple cancers, most notably
Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia(CML).[1] Due
in large part to the development of Gleevec and related drugs having a similar
mechanism of action, the five year survival rate for people with chronic myeloid
leukemia nearly doubled from 31% in 1993 (before Gleevec's 2001 FDA approval) to
59% for those diagnosed between 2003 and 2009.[2] Median survival for
imatinib-treated people with gastrointestinal stromal tumors (GIST) is nearly 5
years compared to 9 to 20 months in the pre-imatinib-era.[3]
Imipenem
亞胺培南
https://en.wikipedia.org/wiki/Imipenem
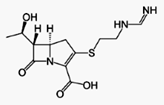
Imipenem (Primaxin) is an intravenous
β-lactam antibiotic discovered by Merck scientists Burton Christensen, William
Leanza, and Kenneth Wildonger in 1980.[1] It was the first member of the
carbapenem class of antibiotics. Carbapenems are highly resistant to the
β-lactamase enzymes produced by many multiple drug-resistant Gram-negative
bacteria,[2] thus play a key role in the treatment of infections not readily
treated with other antibiotics.[3]
It was discovered via a lengthy
trial-and-error search for a more stable version of the natural product
thienamycin, which is produced by the bacterium Streptomyces cattleya.
Thienamycin has antibacterial activity, but is unstable in aqueous solution, so
impractical to administer to patients.[4]Imipenem has a broad spectrum of
activity against aerobic and anaerobic, Gram-positive and Gram-negative
bacteria.[5] It is particularly important for its activity against Pseudomonas
aeruginosa and the Enterococcus species. It is not active against MRSA, however.
Irinotecan hydrochloride
伊立替康
https://en.wikipedia.org/wiki/Irinotecan
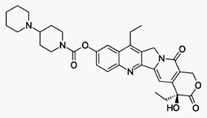
Irinotecan, sold under the brand name
Camptosar, is a medication used for the treatment of cancer. Its main use is in
colon cancer, in particular, in combination with other chemotherapy agents.
Irinotecan prevents DNA from unwinding
by inhibition of topoisomerase 1.[1] In chemical terms, it is a semisynthetic
molecule similar to the natural alkaloid camptothecin.
It is on the WHO
Model List of Essential Medicines, the most important medications needed in a
basic health system.[2] Irinotecan received accelerated approval from the U.S.
Food and Drug Administration (FDA) in 1996 and full approval in 1998.[3][4]
Mechanism of action[edit]
Irinotecan is activated by hydrolysis to SN-38, an inhibitor of topoisomerase I.
This is then inactivated by glucuronidation by uridine diphosphate
glucoronosyltransferase 1A1 (UGT1A1). The inhibition of topoisomerase I by the
active metabolite SN-38 eventually leads to inhibition of both DNA replication
and transcription.
Isoconazole
異康唑
Isoconazol nitrate
https://es.wikipedia.org/wiki/Isoconazol
Isoconazole is an
antifungal drug derived from imidazole that is used in external application to
the skin and mucous membranes. It is usually present in the form of isoconazole
nitrate.
Mechanism of action:
Isoconazole, like other imidazole derivatives, interacts with cytochrome
P450-dependent enzyme systems, interfering with the metabolism of lanosterol
(difficult 14-demethylation) leading to a decrease in ergosterol and,
secondarily, to an accumulation Of anomalous sterols (14-alpha-methylated
sterols). As ergosterol is much more important for the wall of fungi than for
that of human cells, and because of the greater affinity of the former for
azole, Selective action.2 The lack of ergosterol alters the permeability of the
fungi membranes, leading to a disruption of the intracellular organelles and the
ability to divide. Secondarily, the accumulation of anomalous sterols
contributes to cell fragility and death.
Lansoprazole
蘭索拉唑
https://en.wikipedia.org/wiki/Lansoprazole
Lansoprazole is a proton-pump
inhibitor (PPI) which inhibits the stomach's production of gastric acids. It is
manufactured by a number of companies worldwide under several brand names. In
the United States, it was first approved by the Food and Drug Administration
(FDA) in 1995.[1] Prevacid patent protection expired on November 10, 2009.[2][3]
There is not evidence that its effectiveness is different than that of other
PPIs.[4]
Lansoprazole is a proton-pump
inhibitor (PPI) in the same pharmacologic class as omeprazole. Lansoprazole has
been marketed for many years and is one of several PPIs available.[5] It is a
racemic 1:1 mixture of the enantiomers dexlansoprazole (Dexilant, formerly named
Kapidex) and levolansoprazole.[6] Dexlansoprazole is an enantiomerically pure
active ingredient of a commercial drug as a result of the enantiomeric shift.
Lansoprazole's plasma elimination
half-life (1.5 h) is not proportional to the duration of the drug's effects to
the person (i.e. gastric acid suppression).[7] and the effects of the drug last
for over 24 hours after it has been used for a day or more.[8] Lansoprazole,
given nasogastrically, effectively controls intragastric pH and is an
alternative to intravenous pantoprazole in people who are unable to swallow
solid-dose formulations.[9]
Lapatinib Ditosylate
拉帕替尼
二甲苯磺酸鹽
https://en.wikipedia.org/wiki/Lapatinib
Lapatinib (INN), used in the form of
lapatinib ditosylate (USAN) (trade names Tykerb and Tyverb) is an orally active
drug for breast cancer and other solid tumours.[1] It is a dual tyrosine kinase
inhibitor which interrupts the HER2/neu and epidermal growth factor receptor
(EGFR) pathways.[2] It is used in combination therapy for HER2-positive breast
cancer. It is used for the treatment of patients with advanced or metastatic
breast cancer whose tumors overexpress HER2 (ErbB2).[3]
A tyrosine kinase inhibitor (TKI) is a
pharmaceutical drug that inhibits tyrosine kinases. Tyrosine kinases are enzymes
responsible for the activation of many proteins by signal transduction cascades.
The proteins are activated by adding a phosphate group to the protein
(phosphorylation), a step that TKIs inhibit. TKIs are typically used as
anticancer drugs. For example, they have substantially improved outcomes in
chronic myelogenous leukemia.
Latanoprost
拉坦前列腺素
https://en.wikipedia.org/wiki/Latanoprost
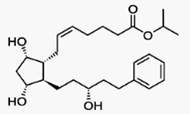
Latanoprost eye
solution is a medication administered into the eyes to control the progression
of glaucoma or ocular hypertension by reducing intraocular pressure (IOP). It is
a prostaglandin analogue (more specifically an analogue of prostaglandin F2α[1])
that lowers the pressure by increasing the outflow of aqueous fluid from the
eyes through the uveoscleral tract.[2] Latanoprost is an isopropyl ester
prodrug, meaning it is inactive until it is hydrolyzed by esterases in the
cornea to the biologically active acid.[3]
Mechanism of action[edit]Like tafluprost and travoprost, latanoprost is an ester
prodrug that is activated to the free acid in the cornea. Also like the related
drugs, latanoprost acid is an analog of prostaglandin F2α that acts as a
selective agonist at the prostaglandin F receptor. Prostaglandins increase the
sclera's permeability to aqueous fluid. So, an increase in prostaglandin
activity increases outflow of aqueous fluid thus lowering intraocular
pressure.[9][10]
Leflunomide
來氟米特
https://en.wikipedia.org/wiki/Leflunomide
Leflunomide (original brand name Arava) is an immunosuppressive
disease-modifying antirheumatic drug (DMARD),[2] used in active
moderate-to-severe rheumatoid arthritis and psoriatic arthritis. It is a
pyrimidine synthesis inhibitor that works by inhibiting dihydroorotate
dehydrogenase.[3]
Dihydroorotate dehydrogenase (DHODH) is an
enzyme that in humans is encoded by the DHODHgene on chromosome 16. The protein
encoded by this gene catalyzes the fourth enzymatic step, the
ubiquinone-mediated oxidation of dihydroorotate to orotate, in de novo
pyrimidine biosynthesis. This protein is a mitochondrial protein located on the
outer surface of the inner mitochondrial membrane(IMM).[1] Inhibitors of this
enzyme are used to treat autoimmune diseases such as rheumatoid arthritis.[2]
Letrozole
來曲唑
https://en.wikipedia.org/wiki/Letrozole
Letrozole (INN, trade name Femara) is an
oral non-steroidal aromatase inhibitor for the treatment of
hormonally-responsive breast cancer after surgery.
Estrogens are produced by the conversion of
androgens through the activity of the aromatase enzyme. Estrogens then bind to
an estrogen receptor, which causes cells to divide.
Letrozole is an aromatase inhibitor.
Letrozole prevents the aromatase from
producing estrogens by competitive, reversible binding to the heme of its
cytochrome P450 unit. The action is specific, and letrozole does not reduce
production of mineralo- or corticosteroids.[citation needed]
Levonorgestrel
左炔諾孕酮
https://en.wikipedia.org/wiki/Levonorgestrel
Levonorgestrel is a manufactured hormone
used in a number of birth control methods.[1] In pill form, sold under the brand
name Plan B among others, it is useful within 120 hours as emergency birth
control. It becomes less effective the longer after sex and only works before
pregnancy has occurred.[1] It is also combined with an estrogen to make combined
oral birth control pill.[2] Within an IUD, sold as Mirena among others, it is
effective for long term prevention of pregnancy.[1] An implantable form of
levonorgestrel is also available in some countries.[3]
Levonorgestrel is an estrane steroid derived
from testosterone and is also known as 17α-ethynyl-18-methyl-19-nortestosterone
or as 17α-ethynyl-18-methylestr-4-en-17β-ol-3-one. Levonorgestrel (levo=left) is
one form of a steroid, norgestrel, that exists in two mirror image left and
right forms (see Chirality (chemistry)). It is the hormonally active
levorotatory enantiomer of the racemic mixture. It is a gonane progestin derived
from 19-nortestosterone.[21]
Its in vitro relative binding affinities at
human steroid hormone receptors are: 323% that of progesterone at the
progesterone receptor, 58% that of testosterone at the androgen receptor, 17%
that of aldosterone at the mineralocorticoid receptor, 7.5% that of cortisol at
the glucocorticoid receptor, and <0.02% that of estradiol at the estrogen
receptor.[22]
If taken together with drugs that induce the
CYP3A4 cytochrome liver enzyme, levonorgestrel may be metabolized faster and may
have lower efficacy.[citation needed]
Lidocaine
利多卡因
Lidocaine HCl
https://en.wikipedia.org/wiki/Lidocaine
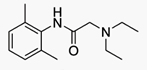
Lidocaine, also known as xylocaine and
lignocaine, is a medication used to numb tissue in a specific area and to treat
ventricular tachycardia.[3][4] It can also be used for nerve blocks. Lidocaine
mixed with a small amount of epinephrine is available to allow larger doses for
numbing, and to make it last longer.[4] When used as an injectable, it typically
begins working within four minutes and lasts for half an hour to three
hours.[4][5] Lidocaine may also be applied directly to the skin for numbing.[4]
Mechanism of action[edit]
Lidocaine alters signal conduction in
neurons by blocking the fast voltage-gated Na+ channels in the neuronal cell
membrane responsible for signal propagation.[34] With sufficient blockage, the
membrane of the postsynaptic neuron will not depolarize and will thus fail to
transmit an action potential. This creates the anaesthetic effect by not merely
preventing pain signals from propagating to the brain, but by stopping them
before they begin. Careful titration allows for a high degree of selectivity in
the blockage of sensory neurons, whereas higher concentrations also affect other
modalities of neuron signaling.
The same principle applies for this drug's
actions in the heart. Blocking sodium channels in the conduction system, as well
as the muscle cells of the heart, raises the depolarization threshold, making
the heart less likely to initiate or conduct early action potentials that may
cause an arrhythmia.[35]
Limaprost Alfadex
利馬前列素
阿法環糊精
https://www.drugs.com/international/limaprost.html
CAS registry number (Chemical Abstracts
Service)
0088852-12-4
Chemical Formula : C22-H36-O5
Molecular Weight : 380
Therapeutic Categories : Vasodilator,
Prostaglandin analogue.
https://en.wikipedia.org/wiki/Prostaglandin_analogue
Synthetic prostaglandin analogues are
molecules which are manufactured to bind to a prostaglandin receptor.
Wider use of prostaglandin analogues is
limited by unwanted side effects and their abortive potential.
Linezolid
利奈唑胺
https://en.wikipedia.org/wiki/Linezolid
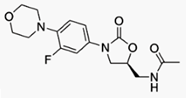
Linezolid is an antibiotic used for the treatment of serious infections caused
by Gram-positive bacteria that are resistant to other antibiotics. Linezolid is
active against most Gram-positive bacteria that cause disease, including
streptococci, vancomycin-resistant enterococci (VRE), and methicillin-resistant
Staphylococcus aureus (MRSA).[1] The main uses are infections of the skin and
pneumoniaalthough it may be use for a variety of other infections..
The oxazolidinones are protein synthesis
inhibitors: they stop the growth and reproduction of bacteria by disrupting
translation of messenger RNA (mRNA) into proteins in the ribosome. Although its
mechanism of action is not fully understood,[90] linezolid appears to work on
the first step of protein synthesis, initiation, unlike most other protein
synthesis inhibitors, which inhibit elongation.[3][54]
It does so by preventing the formation of
the initiation complex, composed of the 30S and 50S subunits of the ribosome,
tRNA, and mRNA. Linezolid binds to the 23S portion of the 50S subunit (the
center of peptidyl transferase activity),[91] close to the binding sites of
chloramphenicol, lincomycin, and other antibiotics. Due to this unique mechanism
of action, cross-resistance between linezolid and other protein synthesis
inhibitors is highly infrequent or nonexistent.[15][46]
Loxoprofen sodium hydrate
洛索丙芬鈉
水合物
https://en.wikipedia.org/wiki/Loxoprofen
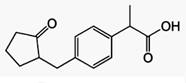
Loxoprofen (INN) is a non-steroidal
anti-inflammatory drug in the propionic acid derivatives group, which also
includes ibuprofen and naproxen among others.
Mechanism of action[edit]
As most NSAIDs, loxoprofen is a
non-selective cyclooxygenase inhibitor, and works by reducing the synthesis of
prostaglandins from arachidonic acid.
Lubiprostone
魯比前列酮
https://en.wikipedia.org/wiki/Lubiprostone
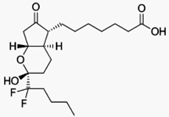
Lubiprostone (rINN, marketed under the trade
name Amitiza among others) is a medicationused in the management of chronic
idiopathic constipation, predominantly irritable bowel syndrome-associated
constipation in women and opioid-induced constipation.
It was initially approved by the U.S. Food
and Drug Administration (FDA) in 2006. It is very expensive as of 2012.[1]
Mechanism of action[edit]
Lubiprostone is a bicyclic fatty acid
derived from prostaglandin E1 that acts by specifically activating ClC-2
chloride channels on the apical aspect of gastrointestinal epithelial cells,
producing a chloride-rich fluid secretion. These secretions soften the stool,
increase motility, and promote spontaneous bowel movements (SBM).
Symptoms of constipation such as pain and
bloating are usually improved within one week, and SBM may occur within one day.
Maxacalcitol Hydrate
馬沙骨化醇
水合物
https://www.medchemexpress.com/DataSheet/Maxacalcitol.html
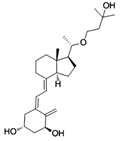
CAS No.: 103909-75-7,
Cat. No.:
HY-32339, MWt:
418.61
Formula: C26H42O4, Purity : >98%, Solubility:
in DMSO > 10 mM
Mechanisms:
Pathways: Vitamin D Related; Target: VD/VDR
Biological Activity: Maxacalcitol
(22-Oxacalcitriol) is non-calcemic vitamin D3 analog and ligand of VDR-like
receptors.
IC50 value: Target:
Maxacalcitol (22-Oxacalcitriol)suppresses
parathyroid hormone (PTH) mRNA expression in vitro and in vivo. Maxacalcitol
exhibits similar effects to calcitriol in osteoblast-like cells.
Maxacalcitol(22-Oxacalcitriol) inhibits tumor growth of osteosarcoma in vitro in
combination with all-trans retinoic acid.
Meclofenamate Sodium
甲氯芬那酸鈉
https://en.wikipedia.org/wiki/Meclofenamic_acid
Meclofenamic acid (meclofenamate sodium,
brand Meclomen) is a drug used for joint, muscular pain, arthritis and
dysmenorrhea.[1] It is a member of the anthranilic acid derivatives (or
fenamate) class of NSAID drugs and was approved by the FDA in 1980.[2] Like
other members of the class, it is a COX inhibitor and prevents formation of
prostaglandins.[3]
Scientists led by Claude Winder from
Parke-Davis invented meclofenamate sodium in 1964, along with fellow members of
the class, mefenamic acid in 1961 and flufenamic acid in 1963.[4]:718
Patents on the drug expired in 1985[5]:295
and several generics were introduced in the US, but as of July 2015 only Mylan
still sold it.[6][7]
It is not widely used in humans as it has a
high rate (30-60%) rate of gastrointestinal side effects.[8]:310 As of 2015 the
cost for a typical course of medication in the United States is 50 to 100
USD.[9]
Mefenamic Acid
甲芬那酸
https://en.wikipedia.org/wiki/Mefenamic_acid
Mefenamic acid is a member of the
anthranilic acid derivatives (or fenamate) class of NSAID drugs, and is used to
treat mild to moderate pain, including menstrual pain, and is sometimes used to
prevent migraines associated with menstruation.[1][2] It is not widely used in
the United States due to its side effects.[3][4]:334
Its name derives from its systematic name,
dimethylphenylaminobenzoic acid. It was discovered and brought to market by
Parke-Davis in the 1960s under brandnames Ponstan, Ponstan Forte, Ponalar,
Ponstyl, and Ponstel. It became generic in the 1980s and is available worldwide
under many brand names.[5] As of 2015 the cost for a typical course of
medication in the United States is more than 200 USD.[6]
Melitracen hydrochloride
米拉明
https://en.wikipedia.org/wiki/Melitracen
Melitracen (Adaptol, Dixeran, Melixeran,
Thymeol, Trausabun) is a tricyclic antidepressant (TCA) marketed in Europe and
Japan by Lundbeck and Takeda, respectively, for the treatment of depression and
anxiety.[1][2][3][4] In addition to single drug preparations, it is also
available as Deanxit, a combination product containing both melitracen and
flupentixol.[5][6][7][8]
The pharmacology of melitracen has not been
properly investigated and is largely unknown, but it is likely to act in a
similar manner to other TCAs. Indeed, melitracen is reported to have imipramine
and amitriptyline-like effects and efficacy against depression and anxiety,
though with improved tolerability and a somewhat faster onset of action.[9][10]
https://en.wikipedia.org/wiki/Tricyclic_antidepressant#PharmacologyThe majority of the TCAs act primarily as
serotonin-norepinephrine reuptake inhibitors (SNRIs) by blocking the serotonin
transporter (SERT) and the norepinephrine transporter (NET), respectively, which
results in an elevation of the synaptic concentrations of these
neurotransmitters, and therefore an enhancement of neurotransmission.[20][21]
Notably, with the sole exception of amineptine, the TCAs have negligible
affinity for the dopamine transporter (DAT), and therefore have no efficacy as
dopamine reuptake inhibitors (DRIs).[20] Both serotonin and norepinephrine have
been highly implicated in depression and anxiety, and it has been shown that
facilitation of their activity has beneficial effects on these mental
disorders.[22]
Meloxicam
美洛昔康
https://en.wikipedia.org/wiki/Meloxicam
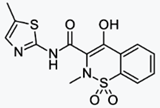
Meloxicam is a nonsteroidal
anti-inflammatory drug (NSAID) with analgesic and fever reducer effects. It is a
derivative of oxicam, closely related to piroxicam, and falls in the enolic acid
group of NSAIDs.[2] It was developed by Boehringer-Ingelheim. Meloxicam starts
to relieve pain about 30–60 minutes after administration.[3]
Mechanism of action[edit]
Main article: Non-steroidal
anti-inflammatory drug
Meloxicam blocks cyclooxygenase (COX), the
enzyme responsible for converting arachidonic acid into prostaglandin H2—the
first step in the synthesis of prostaglandins, which are mediators of
inflammation. Meloxicam has been shown, especially at its low therapeutic doses,
selectively to inhibit COX-2 over COX-1.[1]
Meropenem
美羅培南
Meropenem and Sodium Carbonate
https://en.wikipedia.org/wiki/Meropenem
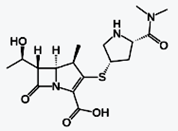
Meropenem is an ultra-broad-spectrum
antibiotic used to treat a wide variety of infections. It is a β-lactam and
belongs to the subgroup of carbapenem, similar to imipenem and ertapenem.
Meropenem was developed by Dainippon
Sumitomo Pharma and patented in 1983.[1][2][3] It gained US FDA approval in July
1996. It penetrates well into many tissues and body fluids, including
cerebrospinal fluid, bile, heart valve, lung, and peritoneal fluid.[4] It was
initially marketed by AstraZeneca under the trade name Merrem.
Mechanism of action[edit]
Meropenem is bactericidal except against
Listeria monocytogenes, where it is bacteriostatic. It inhibits bacterial wall
synthesis like other β-lactam antibiotics. In contrast to other beta-lactams, it
is highly resistant to degradation by β-lactamases or cephalosporinases. In
general, resistance arises due to mutations in penicillin-binding proteins,
production of metallo-β-lactamases, or resistance to diffusion across the
bacterial outer membrane.[5] Unlike imipenem, it is stable to
dehydropeptidase-1, so can be given without cilastatin.
Metaxalone
美他沙酮
https://en.wikipedia.org/wiki/Metaxalone
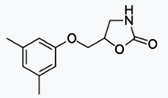
Metaxalone (marketed by King Pharmaceuticals
under the brand name Skelaxin) is a muscle relaxant used to relax muscles and
relieve pain caused by strains, sprains, and other musculoskeletal conditions.
Its exact mechanism of action is not known, but it may be due to general central
nervous system depression. It is considered to be a moderately strong muscle
relaxant, with relatively low incidence of side effects. Skelaxin is available
in an 800 mg scored tablet. Possible side effects include nausea, vomiting,
drowsiness and CNS side effects, such as dizziness, headache, and irritability.
The metabolism of metaxalone involves the
liver cytochrome P450 system. Based on the information in the labeling, patients
receiving metaxalone therapy and physicians prescribing metaxalone are directed
to take precaution when coadministering it with other medications involving the
P450 system.[1][2]
Because of potential for side effects, this
drug is considered high risk in the elderly. As of 2015 the cost for a typical
month of medication in the United States is 100 to 200 USD.[3]
Methocarbamol
甲硫氨醇
Methocarbamol DC 90%
https://en.wikipedia.org/wiki/Methocarbamol
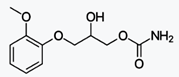
Methocarbamol is a central muscle relaxant
used to treat skeletal muscle spasms. Under the trade name Robaxin, it is
marketed by Actient Pharmaceuticals in the United States and Pfizer in Canada.
The mechanism of action of methocarbamol is currently unknown, but may involve
the inhibition of carbonic anhydrase.[2] The muscle relaxant effects of
methocarbamol are largely attributed to central depressant effects;[3] however,
peripheral effects of methocarbamol to prolong muscle refractory period have
also been reported.
Methylphenidate Hydrochloride
哌甲酯鹽酸鹽
https://en.wikipedia.org/wiki/Methylphenidate
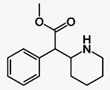
Methylphenidate, sold under various trade
names, Ritalin being one of the most commonly known, is a central nervous system
(CNS) stimulant of the phenethylamine[3] and piperidineclasses that is used in
the treatment of attention deficit hyperactivity disorder (ADHD) and narcolepsy.
The original patent was owned by CIBA, now Novartis Corporation. It was first
licensed by the US Food and Drug Administration (FDA) in 1955 for treating what
was then known as hyperactivity.
Medical use began in 1960; the drug has
become increasingly prescribed since the 1990s, when the diagnosis of ADHD
became more widely accepted.[4][5] Between 2007 and 2012 methylphenidate
prescriptions increased by 50% in the United Kingdom and in 2013 global
methylphenidate consumption increased to 2.4 billion doses, a 66% increase from
the year before. The United States continues to account for more than 80% of
global consumption.[6][7]
ADHD and other similar conditions are
believed to be linked to sub-performance of the dopamine and norepinephrine
functions in the brain, primarily in the prefrontal cortex, responsible for
executive function (e.g., reasoning, inhibiting behaviors, organizing, problem
solving, planning, etc.).[8][9] Methylphenidate's mechanism of action involves
the inhibition of catecholaminereuptake, primarily as a dopamine reuptake
inhibitor. Methylphenidate acts by blocking the dopamine transporter and
norepinephrine transporter, leading to increased concentrations of dopamine and
norepinephrine within the synaptic cleft. This effect in turn leads to increased
neurotransmission of dopamine and norepinephrine.[10] Methylphenidate is also a
weak 5HT1Areceptor agonist.[11]
Metoprolol Succinate
琥珀酸美托洛爾
https://en.wikipedia.org/wiki/Metoprolol
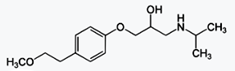
Metoprolol, marketed under the tradename
Lopressor among others, is a selective β1 receptor blocker medication.[3] It is
used to treat high blood pressure, chest pain due to poor blood flow to the
heart, and a number of conditions involving an abnormally fast heart rate. It is
also used to prevent further heart problems after myocardial infarction and to
prevent headaches in those with migraines.[3]
It comes in formulations that can be taken
by mouth or given intravenously. The medication is often taken twice a day.
There is an extended release formulation that is once per day. Metoprolol may be
combined with hydrochlorothiazide in a single tablet.[3]
Common side effects include trouble
sleeping, feeling tired, feeling faint, and abdominal discomfort.[3]Large doses
may cause serious toxicity.[4][5] Risk in pregnancy has not been ruled
out.[3][6] It appears to be safe in breastfeeding.[7] Greater care is required
with use in those with liver problems or asthma.[3] If stopped this should be
done slowly to decrease the risk of further health problems.[3]
Metoprolol was first made in 1969.[8] It is
on the World Health Organization's List of Essential Medicines, the most
important medications needed in a basic health system.[9] It is available as a
generic drug.[3] In 2013, metoprolol was the 19th most prescribed medication in
the United States.[10]
Miconazole Nitrate
咪康唑
https://en.wikipedia.org/wiki/Miconazole
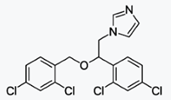
Miconazole, sold under the brand name
Monistat among others, is a antifungal medication used to treat ring worm,
pityriasis versicolor, and yeast infections of the skin or vagina.[1] It is
applied to the skin or vagina as a cream or ointment.[1]
Common side effects include itchiness or
irritation of the area in which it was applied.[1] Use in pregnancy is believed
to be safe for the baby.[2] Miconazole is in the imidazole family of
medications. It works by decreasing the ability of fungi to make ergosterol, an
important part its cell membrane.[1]
Miconazole was patented in 1968 and approved
for medical use in 1971.[3] It is on the World Health Organization's List of
Essential Medicines, the most important medications needed in a basic health
system.[4]
Miconazole, itraconazole, and clotrimazole
work in a different way, inhibiting synthesis of ergosterol from lanosterol by
interfering with 14α-demethylase . Ergosterol is a smaller molecule than
lanosterol; it is synthesized by combining two molecules of farnesyl
pyrophosphate, a 15-carbon-long terpenoid, into lanosterol, which has 30
carbons. Then, two methyl groups are removed, making ergosterol. The "azole"
class of antifungal agents inhibit the enzyme that performs these demethylation
steps in the biosynthetic pathway between lanosterol and ergosterol.
Mifepristone
米非司酮
https://en.wikipedia.org/wiki/Mifepristone
Mifepristone, also known as RU-486, is a
medication typically used with misoprostol to bring about an abortion.[1] This
combination is more than 95% effective during the first 50 days of pregnancy. It
is also effective in the second trimester of pregnancy.[2][3] Two weeks after
use effectiveness should be verified. It is taken by mouth.[1]
Pharmacology[edit]
It is a synthetic, steroidal antiprogestogen
(IC50 = 0.025 nM for the PR), as well as an antiglucocorticoid (IC50 = 2.2 nM
for the GR) and antiandrogen (IC50 = 10 nM for the AR) to a much lesser
extent.[32] It is a 19-norsteroid with substitutions at positions C11 and C17
(17β-hydroxy-11β-(4-(dimethylamino)phenyl)-17α-(1-propynyl)estra-4,9-dien-3-one),
which antagonizes cortisol action competitively at the receptor
level.[33]Mifepristone is a low-efficacy partial agonist of the progesterone
receptor. It is also a glucocorticoid receptor antagonist to a lesser extent.
In the presence of progesterone,
mifepristone acts as a competitive progesterone receptor antagonist (in the
absence of progesterone, mifepristone acts as a partial agonist). Mifepristone
is a 19-nor steroid with a bulky p-(dimethylamino)phenyl substituent above the
plane of the molecule at the 11β-position responsible for inducing or
stabilizing an inactive receptor conformation and a hydrophobic 1-propynyl
substituent below the plane of the molecule at the 17α-position that increases
its progesterone receptor binding affinity.[34][35][36]
Miltefosine
米替福新
https://en.wikipedia.org/wiki/Miltefosine
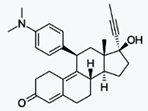
Miltefosine, sold under the trade name
Impavido among others, is a medication mainly used to treat leishmaniasis and
free-living amoeba infections such as Naegleria fowleri.[1] This includes
leishmaniasis of the cutaneous, visceral, and mucosal types.[3] It may be used
together with liposomal amphotericin B or paromomycin.[4] It is taken by
mouth.[3]
Common side effects include vomiting,
abdominal pain, fever, headaches, and decreased kidney function. More severe
side effects may include Stevens-Johnson syndrome or low blood platelets. Use
during pregnancy appears to cause harm to the baby and use during breastfeeding
is not recommended. How it works is not entirely clear.[1]
Miltefosine was first made in the early
1980s and studied as a treatment for cancer.[5] A few years later it was found
to be useful for leishmaniasis and was approved for this use in 2002 in
India.[6] It is on the World Health Organization's List of Essential Medicines,
the most important medications needed in a basic health system.[7] In the
developing world a course of treatment costs 65 to 150 USD. In the developed
world treatment may be 10 to 50 times greater.[4]
Mechanism of action[edit]
Miltefosine primarily acts on Leishmania by
affecting the species promastigote and amastigote stages.[23] Miltefosine exerts
its activity by interacting with lipids, inhibiting cytochrome c oxidase and
causing apoptosis-like cell death.[24] This may affect membrane integrity and
mitochondrial function of the parasite.
Misoprostol
米索前列醇
Misoprostol 1% HPMC Dispersion
Misoprostol-HPMC 1% Dispersion
https://en.wikipedia.org/wiki/Misoprostol
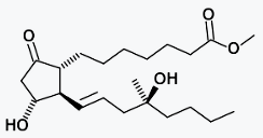
Misoprostol, sold under the brandname
Cytotec among others, is a medication used to start labor, cause an abortion,
prevent and treat stomach ulcers, and treat postpartum bleeding due to poor
contraction of the uterus.[1] For abortions it is often used with mifepristone
or methotrexate.[2] By itself effectiveness for this purpose is between 66% and
90%.[3] It is taken either by mouth, under the tongue, or placed in the
vagina.[2][4]
Common side effects include diarrhea and
abdominal pain. It is pregnancy category X meaning that it is known to result in
negative outcomes for the baby if taken during pregnancy. Uterine rupture may
occur. It is a prostaglandin analogue — specifically, a synthetic prostaglandin
E1 (PGE1).[1]
Misoprostol was developed in 1973.[5] It is
on the World Health Organization's List of Essential Medicines, the most
important medications needed in a basic health system.[6] It is available as a
generic medication.[1] The wholesale cost in the developing world is about 0.36
to 2.00 USD a dose.[7] A months supply to treat stomach ulcers in the United
States is between 100 and 200 USD.[8] The same costs between 30 and 55 EUR in
Europe.[9]
Montelukast Sodium
孟魯司特鈉
https://en.wikipedia.org/wiki/Montelukast
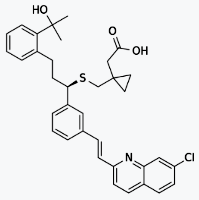
Montelukast (trade name Singulair) is a
leukotriene receptor antagonist (LTRA) used for the maintenance treatment of
asthma and to relieve symptoms of seasonal allergies.[2][3] Montelukast comes as
a tablet, a chewable tablet, flash tablet and granules to take by mouth.[4]
Montelukast is usually taken once a day with or without food.[4] Montelukast is
a CysLT1 antagonist; it blocks the action of leukotriene D4 (and secondary
ligands LTC4 and LTE4) on the cysteinyl leukotriene receptor CysLT1 in the lungs
and bronchial tubes by binding to it. This reduces the bronchoconstriction
otherwise caused by the leukotriene and results in less inflammation.
Because of its mechanism of action, it is
not useful in the treatment of acute asthma attacks.
Another leukotriene receptor antagonist is
zafirlukast (Accolate). Zileuton (Zyflo), an asthma drug, blocks leukotriene
synthesis by inhibiting 5-lipoxygenase, an enzyme of the eicosanoid synthesis
pathway.[5]
The Mont in Montelukast stands for Montreal,
the place where Merck developed the drug.[6]
Mupirocin
莫匹羅星
https://en.wikipedia.org/wiki/Mupirocin
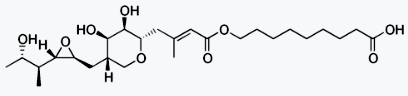
Mupirocin, sold under the brand name
Bactroban among others, is an antibiotic useful against superficial skin
infections such as impetigo or folliculitis.[1][2] It may also be used to get
rid of methicillin-resistant S. aureus (MRSA) when present in the nose without
symptoms.[1] Due to concerns of developing resistance, use for greater than ten
days is not recommended.[2] It is used as a cream or ointment applied to the
skin.[1]
Common side effects include itchiness and
rash at the site of application, headache, and nausea. Long term use may result
in increased growth of fungi. Use during pregnancy and breastfeeding appear to
be safe.[1] Mupirocin is in the carbolic acid class of medications.[3]It works
by blocking the making of protein by the bacterial which usually results in
bacterial death.[1]
Mupirocin was initially isolated in 1971
from Pseudomonas fluorescens.[4] It is on the World Health Organization's List
of Essential Medicines, the most important medications needed in a basic health
system.[5]
Mechanism of action[edit]
Mupirocin reversibly binds to the isoleucyl
t-RNA synthetase in Staphylococcus aureus and Streptococcus, resulting in
inhibition of protein synthesis. DNA and cell wall formation are also negatively
impacted to a lesser degree.[15] The inhibition of RNA synthesis was shown to be
a protective mechanism in response to a lack of one amino acid, isoleucine.[16]
In vivo studies in Escherichia coli demonstrated that pseudomonic acid inhibits
isoleucine t-RNA synthetase (IleRS).[8] This mechanism of action is shared with
furanomycin, an analog of isoleucine.[17]
Mycophenolate mofetil
黴酚酸酯
https://en.wikipedia.org/wiki/Mycophenolic_acid
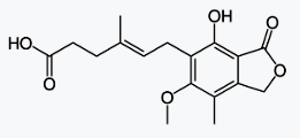
Mycophenolic acid, less accurately called
mycophenolate, is an immunosuppressant drug used to prevent rejection in organ
transplantation. It inhibits an enzyme needed for the growth of T cells and B
cells. It was initially marketed as the prodrug mycophenolate mofetil (MMF) to
improve oral bioavailability. More recently, the salt mycophenolate sodium has
also been introduced. Mycophenolate mofetil is marketed under the trade name
CellCept and mycophenolate sodium as Myfortic.
Discovered by an Italian medical scientist
Bartolomeo Gosio in 1893, mycophenolic acid was the first antibiotic to be
synthesised in pure and crystalline form. But its medical application was
forgotten until two American scientists C.L. Alsberg and O.M. Black
resynthesised it in 1912, and gave its chemical name. It was eventually found to
be a broad-spectrum acting drug having antiviral, antifungal, antibacterial,
anticancer, and antipsoriasis properties.[3] The clinically usable drug Cellcept
was developed by South African geneticist Anthony Allison and his wife Elsie M.
Eugui. It was first approved by the US Food and Drug Administration on 3 May
1995 for use in kidney transplantation.[4]
Naftopidil
萘夫地爾
https://en.wikipedia.org/wiki/Naftopidil
Naftopidil (INN, marketed under the brand
name Flivas) is a drug used in benign prostatic hypertrophy which acts as a
selective α1-adrenergic receptor antagonist or alpha blocker.[1]
Nicametate Citrate
枸橼酸烟胺乙酯https://pubchem.ncbi.nlm.nih.gov/compound/Nicametate_citrate#section=Top
Chemical Names: Nicametate citrate; Euclidan
Molecular Formula: C18H26N2O9,
Molecular Weight: 414.411 g/mol
Vasodilator Agents: Drugs used to cause
dilation of the blood vessels.
Nicotinamide
煙酰胺
https://en.wikipedia.org/wiki/Nicotinamide
Nicotinamide, (/ˌnɪkəˈtɪnəmaɪd/) also known
as niacinamide,[2][3] NAA, and nicotinic amide, is the amide of nicotinic acid
(vitamin B3 / niacin).[2][3] Nicotinamide is a water-soluble vitamin and is part
of the vitamin B group. Nicotinic acid, also known as niacin, is converted to
nicotinamide in vivo, and, though the two are identical in their vitamin
functions, nicotinamide does not have the same pharmacological and toxic effects
of niacin, which occur incidental to niacin's conversion. Thus nicotinamide does
not reduce cholesterol or cause flushing,[4] although nicotinamide may be toxic
to the liver at doses exceeding 3 g/day for adults.[5] In cells, niacin is
incorporated into nicotinamide adenine dinucleotide (NAD) and nicotinamide
adenine dinucleotide phosphate (NADP), although the pathways for nicotinic acid
amide and nicotinic acid are very similar. NAD+ and NADP+ are coenzymes in a
wide variety of enzymatic oxidation-reduction reactions.[6] Commercial
production of niacin and niacinamide (several thousand tons annually) is by
hydrolysis or aminolysis of 3-cyanopyridine (nicotinonitrile).[7]
Small intestinal bacterial overgrowth is one
known cause of nicotinamide deficiency.
Olopatadine HCl
奧洛他定
https://en.wikipedia.org/wiki/Olopatadine
Olopatadine hydrochloride is an
antihistamine (as well as anticholinergic and mast cell stabilizer), sold as a
prescription eye drop manufactured by Alcon in one of three strengths: 0.7%
solution or Pazeo in the US, 0.2% solution or Pataday (also called Patanol S in
some countries), and 0.1% or Patanol (also called Opatanol in some countries).
It is used to treat itching associated with allergic conjunctivitis (eye
allergies). A decongestant nasal spray formulation is sold as Patanase, which
was approved by the FDA on April 15, 2008.[1] It is also available as an oral
tablet in Japan under the tradename Allelock, manufactured by Kyowa Hakko
Kogyo.[2]
It should not be used to treat irritation
caused by contact lenses. The usual dose for Patanol is 1 drop in each affected
eye 2 times per day, with 6 to 8 hours between doses. Both Pazeo and Pataday are
dosed 1 drop in each eye daily.
There is potential for Olopatadine as a
treatment modality for steroid rebound (red skin syndrome).[3]
Olopatadine was developed by Kyowa Hakko
Kogyo.[4]
Omeprazole
奧美拉唑
https://en.wikipedia.org/wiki/Omeprazole
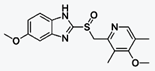
Omeprazole, sold under the brand names
Prilosec and Losec among others, is a medication used in the treatment of
gastroesophageal reflux disease, peptic ulcer disease, and Zollinger–Ellison
syndrome.[1] It is also used to prevent upper gastrointestinal bleeding in
people who are at high risk.[1] It can be taken by mouth or injected into a
vein.[1][4]
Common side effects include nausea,
vomiting, headaches, and increased intestinal gas. Serious side effects may
include Clostridium difficile colitis, an increased risk of pneumonia, an
increased risk of bone fractures, and the potential of masking stomach cancer.
It is unclear if it is safe for use in pregnancy. Omeprazole is a proton pump
inhibitor and as such blocks the release of stomach acid.[1]
Omeprazole was discovered in 1979.[5] It is
on the World Health Organization's List of Essential Medicines, the most
important medications needed in a basic health system.[6] It is available as a
generic medication.[1]
Ondansetron HCl
昂丹司瓊
https://en.wikipedia.org/wiki/Ondansetron
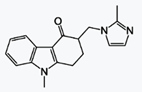
Ondansetron, marketed under the brand name
Zofran, is a medication used to prevent nausea and vomiting caused by cancer
chemotherapy, radiation therapy, or surgery.[1] It is also useful in
gastroenteritis.[2][3] It has little effect on vomiting caused by motion
sickness.[4] It can be given by mouth, by injection into a muscle or into a
vein.[1]
Common side effects include diarrhea,
constipation, headache, sleepiness, and itchiness. Serious side effects include
QT prolongation and severe allergic reaction. It appears to be safe during
pregnancy but has not been well studied in this group. It is a serotonin 5-HT3
receptor antagonist.[1] It does not have any effect on dopamine receptors or
muscarinic receptors.[5]
Ondansetron was first used medically in
1990.[6] It is on the WHO Model List of Essential Medicines, the most important
medications needed in a basic health system.[7] It is available as a generic
medication.[1] The wholesale cost of the injectable form in the developing world
is about 0.10 to 0.76 USD per dose.[8] In the United States it costs about 1.37
USD per tablet.[1]
Orciprenaline Sulphate
奧西那林
https://en.wikipedia.org/wiki/Orciprenaline
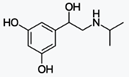
Orciprenaline (INN), also known as
metaproterenol (USAN), is a bronchodilator used in the treatment of
asthma.[1][2] Orciprenaline is a moderately selective β2 adrenergic receptor
agonist that stimulates receptors of the smooth muscle in the lungs, uterus, and
vasculature supplying skeletal muscle, with minimal or no effect on α adrenergic
receptors. The pharmacologic effects of β adrenergic agonist drugs, such as
orciprenaline, are at least in part attributable to stimulation through β
adrenergic receptors of intracellular adenylyl cyclase, the enzyme which
catalyzes the conversion of ATP to cAMP. Increased cAMP levels are associated
with relaxation of bronchial smooth muscle and inhibition of release of
mediators of immediate hypersensitivity from many cells, especially from mast
cells.
Orlistat
奧利司他
https://en.wikipedia.org/wiki/Orlistat
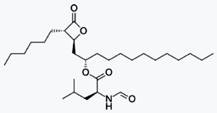
Orlistat is a drug designed to treat
obesity. It is marketed as a prescription drug under the trade name Xenical by
Roche in most countries, and is sold over-the-counter as Alli[2] by
GlaxoSmithKline in the United Kingdom and the United States.[3] Its primary
function is preventing the absorption of fats from the human diet by acting as a
lipase inhibitor, thereby reducing caloric intake. It is intended for use in
conjunction with a healthcare provider-supervised reduced-calorie diet.[4]
Orlistat is the saturated derivative of
lipstatin, a potent natural inhibitor of pancreatic lipases isolated from the
bacterium Streptomyces toxytricini.[5] However, due to its relative simplicity
and stability, orlistat was chosen over lipstatin for development as an
anti-obesity drug.[6]
Oteracil Potassium
氧嗪酸鉀https://en.wikipedia.org/wiki/Tegafur/gimeracil/oteracil
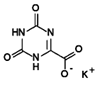
Tegafur/gimeracil/oteracil
The combination drug
tegafur/gimeracil/oteracil (trade name Teysuno, and TS-1 in Japan[1]), also
known as S-1,[2] is used for the treatment of advanced gastric cancer.[3] It is
labelled for use in combination with cisplatin in many European countries, and
for head and neck cancer, colorectal cancer, non–small-cell lung, breast,
pancreatic, and biliary tract cancers in several countries in Asia.[4]:213 It
has not been approved by the FDA.[4]:213
It is also being developed for the treatment
of hepatocellular carcinoma.[5] and has activity in esophageal,(Perry Chapter
33) breast,[citation needed] cervical,[citation needed] and colorectal
cancer.[6]
Mechanism of action[edit]
Tegafur is the actual chemotherapeutic
agent. It is a prodrug of the active substance fluorouracil (5-FU).
Gimeracil inhibits the degradation of
fluorouracil by reversibly blocking a dehydrogenase enzyme. This results in
higher 5-FU levels and a prolonged half-life of the substance.
Oteracil mainly stays in the gut because of
its low permeability, where it reduces the production of 5-FU by blocking the
enzyme orotate phosphoribosyltransferase. Lower 5-FU levels in the gut result in
a lower gastrointestinal toxicity.
Oxazolam
噁唑崙
https://en.wikipedia.org/wiki/Oxazolam
Oxazolam is a drug that is a benzodiazepine
derivative. It has anxiolytic, anticonvulsant, sedative, and skeletal muscle
relaxant properties. It is a prodrug for desmethyldiazepam.[1]
Oxethazaine
奧昔卡因
https://en.wikipedia.org/wiki/Oxetacaine
Oxetacaine (INN, also known as oxethazaine)
is a potent local anesthetic. It is administered orally (usually in combination
with an antacid) for the relief of pain associated with peptic ulcer disease or
esophagitis. It is also used topically in the management of hemorrhoid pain.
Oral oxetacaine preparations are available in several countries, including
India, South Africa, Japan and Brazil, but not the United States.
Unlike most local anesthetics, oxetacaine
does not break down under strongly acidic conditions.[1]
https://en.wikipedia.org/wiki/Local_anesthetic
Mechanism of action[edit]
All LAs are membrane-stabilizing drugs; they
reversibly decrease the rate of depolarization and repolarization of excitable
membranes (like nociceptors). Though many other drugs also have
membrane-stabilizing properties, not all are used as LAs (propranolol, for
example). LA drugs act mainly by inhibiting sodium influx through
sodium-specific ion channels in the neuronal cell membrane, in particular the
so-called voltage-gated sodium channels. When the influx of sodium is
interrupted, an action potential cannot arise and signal conduction is
inhibited. The receptor site is thought to be located at the cytoplasmic (inner)
portion of the sodium channel. Local anesthetic drugs bind more readily to
sodium channels in an activated state, thus onset of neuronal blockade is faster
in rapidly firing neurons. This is referred to as state-dependent blockade.
LAs are weak bases and are usually
formulated as the hydrochloride salt to render them water soluble. At a pH equal
to the protonated base's pKa, the protonated (ionized) and un-protonated
(unionized) forms of the molecule exist in equal molar amounts, but only the
un-protonated base diffuses readily across cell membranes. Once inside the cell,
the local anesthetic will be in equilibrium, with the formation of the
protonated (ionized form), which does not readily pass back out of the cell.
This is referred to as "ion-trapping". In the protonated form, the molecule
binds to the LA binding site on the inside of the ion channel near the
cytoplasmic end. Most LAs work on the internal surface of the membrane - the
drug has to penetrate the cell membrane, which is achieved best in the
non-ionized form.
Paclitaxel
紫杉醇
https://en.wikipedia.org/wiki/Paclitaxel
Paclitaxel (PTX), sold under the brand name
Taxol among others, is a chemotherapy medication used to treat a number of types
of cancer. This includes ovarian cancer, breast cancer, lung cancer, Kaposi
sarcoma, cervical cancer, and pancreatic cancer. It is given by injection into a
vein.[2] There is also an albumin bound formulation.[2]
Common side effects include hair loss, bone
marrow suppression, numbness, allergic reactions, muscle pains, and diarrhea.
Other serious side effects include heart problems, increased risk of infection,
and lung inflammation. Use during pregnancy may result in harm to the baby.[2]
Paclitaxel is in the taxane family of medications.[3] It works by interference
with the normal function of microtubules during cell division.[2]
Paclitaxel was first isolated in 1971 from
the Pacific yew and approved for medical use in 1993.[4][5] It is on the World
Health Organization's List of Essential Medicines, the most important medication
needed in a basic health system.[6] The wholesale cost in the developing world
is about 7.06 to 13.48 USD per 100 mg vial.[7] This amount in the United Kingdom
costs the NHS about 66.85 pounds.[8] It is now manufactured by cell culture.[5]
Mechanism of action[edit]
Paclitaxel is one of several cytoskeletal
drugs that target tubulin. Paclitaxel-treated cells have defects in mitotic
spindle assembly, chromosome segregation, and cell division. Unlike other
tubulin-targeting drugs such as colchicine that inhibit microtubule assembly,
paclitaxel stabilizes the microtubule polymer and protects it from disassembly.
Chromosomes are thus unable to achieve a metaphase spindle configuration. This
blocks the progression of mitosis and prolonged activation of the mitotic
checkpoint triggers apoptosis or reversion to the G-phase of the cell cycle
without cell division.[18][19]
The ability of paclitaxel to inhibit spindle
function is generally attributed to its suppression of microtubule dynamics,[20]
but recent studies have demonstrated that suppression of dynamics occurs at
concentrations lower than those needed to block mitosis. At the higher
therapeutic concentrations, paclitaxel appears to suppress microtubule
detachment from centrosomes, a process normally activated during
mitosis.[21]Paclitaxel binds to beta-tubulin subunits of microtubules.[22]
p-Aminomethylbenzoic acid
對氨基甲基苯甲酸https://en.wikipedia.org/wiki/Aminomethylbenzoic_acid
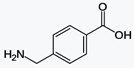
Aminomethylbenzoic acid (more precisely,
4-aminomethylbenzoic acid or p-aminomethylbenzoic acid, PAMBA) is an
antifibrinolytic.
https://en.wikipedia.org/wiki/Antifibrinolytic
Antifibrinolytics, such as aminocaproic acid
(ε-aminocaproic acid) and tranexamic acid are used as inhibitors of
fibrinolysis.[1] These lysine-like drugs interfere with the formation of the
fibrinolytic enzyme plasmin from its precursor plasminogen by plasminogen
activators (primarily t-PA and u-PA) which takes place mainly in lysine rich
areas on the surface of fibrin. These drugs block the binding sites of the
enzymes or plasminogen respectively and thus stop plasmin formation.
They are used in menorrhagia and bleeding
tendency due to various causes. Their application may be beneficial in patients
with hyperfibrinolysis because they arrest bleeding rapidly if the other
components of the haemostatic system are not severely affected. This may help to
avoid the use of blood products such as fresh frozen plasma (FFP) with its
associated risks of infections or anaphylactic reactions.
In 2010, the CRASH-2 trial showed that the
antifibrinolytic drug tranexamic acid safely reduces mortality in bleeding
trauma patients.[2]
The antifibrinolytic drug aprotinin was
abandoned after identification of major side effects, especially on kidney.
The indication for use of antifibrinolytic
drugs is made with various methods. The most rapid and suitable one is
thromboelastometry (TEM) in whole blood, which is even possible in patients on
heparin. With various assays, an enhanced fibrinolysis becomes visible in the
curve signature (TEMogram) and from the calculated values, e.g. the maximum
lysis parameter. A special test for the identification of increased fibrinolysis
(APTEM) compares the TEM in the absence or presence of the fibrinolysis
inhibitor aprotinin. In severe cases of activated fibrinolysis, this assay
confirms the syndrome already in less than 15 min during the early phases of
clot formation [3]
Paricalcitol
帕立骨化醇
https://en.wikipedia.org/wiki/Paricalcitol
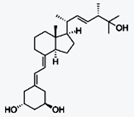
Paricalcitol (chemically it is
19-nor-1,25-(OH)2-vitamin D2. Marketed by Abbott Laboratories under the trade
name Zemplar) is a drug used for the prevention and treatment of secondary
hyperparathyroidism (excessive secretion of parathyroid hormone) associated with
chronic renal failure. It is an analog of 1,25-dihydroxyergocalciferol, the
active form of vitamin D2 (ergocalciferol).
Mechanism of action[edit]
Like 1,25-dihydroxyergocalciferol,
paricalcitol acts as an agonist at the vitamin D receptor and thereby lowers
parathyroid hormone levels in the blood.[1]
Pazopanib Hydrochloride
鹽酸帕唑帕尼
https://en.wikipedia.org/wiki/Pazopanib
Pazopanib (trade name Votrient) is a potent
and selective multi-targeted receptor tyrosine kinase inhibitor that blocks
tumour growth and inhibits angiogenesis. It has been approved for renal cell
carcinoma and soft tissue sarcoma by numerous regulatory administrations
worldwide.[3][4][5][6]
Mechanism of action[edit]
It is a multikinase inhibitor, with c-KIT,
FGFR, PDGFR and VEGFR being amongst the inhibited
enzymes.[2][12][15][16][17][18]
Pemetrexed Disodium Hemipentahydrate
培美曲塞半水合二鈉
https://en.wikipedia.org/wiki/Pemetrexed
Pemetrexed (brand name Alimta) is a
chemotherapy drug manufactured and marketed by Eli Lilly and Company. Its
indications are the treatment of pleural mesothelioma and non-small cell lung
cancer.
Mechanism of action[edit]
Pemetrexed is chemically similar to folic
acid and is in the class of chemotherapy drugs called folate antimetabolites. It
works by inhibiting three enzymes used in purine and pyrimidine
synthesis—thymidylate synthase (TS), dihydrofolate reductase (DHFR), and
glycinamide ribonucleotide formyltransferase[16][17](GARFT). By inhibiting the
formation of precursor purine and pyrimidine nucleotides, pemetrexed prevents
the formation of DNA and RNA, which are required for the growth and survival of
both normal cells and cancer cells.
Pentobarbital Sodium
戊巴比妥鈉
https://en.wikipedia.org/wiki/Pentobarbital
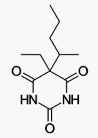
Pentobarbital (US English) or pentobarbitone
(UK English) is a short-acting barbiturate. Pentobarbital can occur as both a
free acid and as salts of elements such as sodium and calcium. The free acid is
only slightly soluble in water and ethanol.[1][2]
One brand name for this drug is Nembutal,
coined by John S. Lundy, who started using it in 1930, from the structural
formula of the sodium salt—Na (sodium) + ethyl + methyl + butyl + al (common
suffix for barbiturates).[3] Nembutal is trademarked and manufactured by the
Danish pharmaceutical company Lundbeck, and is the only injectable form of
pentobarbital approved for sale in the United States.[4]
In high doses, pentobarbital causes death by
respiratory arrest. In the United States, the drug has been used for executions
of convicted criminals. Lundbeck (one of many manufacturers) does not permit its
sale to prisons or corrections departments to carry out the death penalty.[5]
Mechanism of action[edit]
https://en.wikipedia.org/wiki/Barbiturate#Mechanism_of_action
Barbiturates act as positive allosteric
modulators, and at higher doses, as agonists of GABAA receptors.[20] GABA is the
principal inhibitory neurotransmitter in the mammalian central nervous system
(CNS). Barbiturates bind to the GABAA receptor at multiple homologous
transmembrane pockets located at subunit interfaces,[21] which are binding sites
distinct from GABA itself and also distinct from the benzodiazepine binding
site. Like benzodiazepines, barbiturates potentiate the effect of GABA at this
receptor. In addition to this GABAergic effect, barbiturates also block AMPA and
kainate receptors, subtypes of ionotropic glutamate receptor. Glutamate is the
principal excitatory neurotransmitter in the mammalian CNS. Taken together, the
findings that barbiturates potentiate inhibitory GABAA receptors and inhibit
excitatory AMPA receptors can explain the superior CNS-depressant effects of
these agents to alternative GABA potentiating agents such as benzodiazepines and
quinazolinones. At higher concentration, they inhibit the Ca2+-dependent release
of neurotransmitters such as glutamate via an effect on P/Q-type
voltage-dependent calcium channels.[22] Barbiturates produce their
pharmacological effects by increasing the duration of chloride ion channel
opening at the GABAA receptor (pharmacodynamics: This increases the efficacy of
GABA), whereas benzodiazepines increase the frequency of the chloride ion
channel opening at the GABAA receptor (pharmacodynamics: This increases the
potency of GABA). The direct gating or opening of the chloride ion channel is
the reason for the increased toxicity of barbiturates compared to
benzodiazepines in overdose.[23][24]
Further, barbiturates are relatively
non-selective compounds that bind to an entire superfamily of ligand-gated ion
channels, of which the GABAAreceptor channel is only one of several
representatives. This superfamily of ion channels includes the neuronal nACh
receptor channel, the 5-HT3receptor channel, and the glycine receptor channel.
However, while GABAA receptor currents are increased by barbiturates (and other
general anaesthetics), ligand-gated ion channels that are predominantly
permeable for cationic ions are blocked by these compounds. For example,
neuronal nAChR channels are blocked by clinically relevant anaesthetic
concentrations of both thiopental and pentobarbital.[25] Such findings implicate
(non-GABA-ergic) ligand-gated ion channels, e.g. the neuronal nAChR channel, in
mediating some of the (side) effects of barbiturates.[26]This is the mechanism
responsible for the (mild to moderate) anesthetic effect of barbiturates in high
doses when used in anesthetic concentration
Phenylephrine bitartrate
去氧腎上腺素
酒石酸鹽
Phenylephrine HCl
Phenylephrine Hydrochloride
https://en.wikipedia.org/wiki/Phenylephrine
Phenylephrine is a selective α1-adrenergic
receptor agonist of the phenethylamine class used primarily as a decongestant,
as an agent to dilate the pupil, and to increase blood pressure. Phenylephrine
is marketed as an alternative for the decongestant pseudoephedrine, although
clinical trials show phenylephrine, taken orally at the recommended dose, to be
no more effective than placebo for allergy relief.[1][2] Phenylephrine can also
cause a decrease in heart rate through reflex bradycardia.[3]
Phenylpropanolamine Hydrochloride
苯丙醇胺https://en.wikipedia.org/wiki/Phenylpropanolamine
Phenylpropanolamine (BAN and INN; PPA,
β-hydroxyamphetamine), also known as the stereoisomers norephedrine,
norpseudoephedrine, and cathine, is a psychoactive drug of the phenethylamine
and amphetamine chemical classes which is used as a stimulant, decongestant, and
anorectic agent.[1] It is commonly used in prescription and over-the-counter
cough and cold preparations. In veterinary medicine, it is used to control
urinary incontinence in dogs under trade names Propalin and Proin.
In the United States, PPA is no longer sold
due to a purported increased risk of stroke in younger women. In a few countries
in Europe, however, it is still available either by prescription or sometimes
over-the-counter. In Canada, it was withdrawn from the market on 31 May 2001.[2]
In India human use of PPA and its formulations was banned on 10 February
2011,[3] but the ban was overturned by the judiciary in September 2011.[4]Pharmacology[edit]
Phenylpropanolamine acts as an
alpha-adrenergic receptor and beta-adrenergic receptor agonist as well as a
dopamine receptor D1 partial agonist.[5]
Many sympathetic hormones and
neurotransmitters are based on the phenethylamine skeleton, and function
generally in "fight or flight" type responses, such as increasing heart rate,
blood pressure, dilating the pupils, increased energy, drying of mucous
membranes, increased sweating, and a significant number of additional effects.
Phenyltoloxamine Citrate
苯基托沙胺
https://en.wikipedia.org/wiki/Phenyltoloxamine
Phenyltoloxamine is an antihistamine with
sedative and analgesic effects. It is a member of the ethanolamine class of
antihistaminergic agents and an anticholinergic.
Common use[edit]
Phenyltoloxamine is widely used in
preparations as an enhancing agent for some analgesics and antitussives
(acetaminophen, dihydrocodeine, codeine, hydrocodone). It is widely used in
certain parts of the world as cough suppressant usually with codeine, and
sometimes by itself or in addition to dextromethorphan as it, like
diphenhydramine, possesses antitussive action of its own and is particularly
useful in semi-productive coughs because of its moderate drying action.
https://en.wikipedia.org/wiki/Antihistamine
An antihistamine is a type of pharmaceutical
drug that opposes the activity of histamine receptors in the body.[1]
Antihistamines are subclassified according to the histamine receptor that they
act upon: the two largest classes of antihistamines are H1-antihistamines and
H2-antihistamines. Antihistamines that target the histamine H1-receptor are used
to treat allergic reactions in the nose (e.g., itching, runny nose, and
sneezing) as well as for insomnia. They are sometimes also used to treat motion
sickness or vertigo caused by problems with the inner ear. Antihistamines that
target the histamine H2-receptor are used to treat gastric acid conditions
(e.g., peptic ulcers and acid reflux). H1-antihistamines work by binding to
histamine H1 receptors in mast cells, smooth muscle, and endothelium in the body
as well as in the tuberomammillary nucleus in the brain; H2-antihistamines bind
to histamine H2 receptors in the upper gastrointestinal tract, primarily in the
stomach.
Pipethanate Ethobromide
http://www.druginfosys.com/drug.aspx?drugcode=2164&type=1#Shortcuts
Overview
Pipethanate ethobromide is an antimuscarinic
with actions similar to those of atropine.
Categories
4 Antidotes and other substances used in
poisonings
4.2 Specific antidotes
4.2.3 Organophosphate and carbamate
poisoning
17 Gastrointestinal drugs
17.5 Antispasmodics
Side Effects
The severe or irreversible adverse effects
of Pipethanate ethobromide, which give rise to further complications include
Deaths.
The signs and symptoms that are produced
after the acute overdosage of Pipethanate ethobromide include Nausea, Vomiting,
Confusion, Hallucinations, Ataxia, CNS stimulation, Incoordination, Rashes,
Paranoid psychosis, Increased respiration rate, Excitement.
Potassium Cresolsulfonate
煤溜油酚磺酸鉀http://www.chemicalbook.com/ChemicalProductProperty_CN_CB11104948.htm
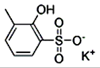
Pramoxine HCl
普莫林
https://en.wikipedia.org/wiki/Pramocaine
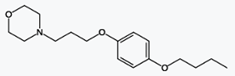
Pramocaine (INN and BAN, also known as
pramoxine or pramoxine HCI) is a topical anesthetic discovered at Abbott
Laboratories in 1953[1] and used as an antipruritic. During research and
development, pramocaine hydrochloride stood out among a series of alkoxy aryl
alkamine ethers as an especially good topical local anesthetic agent.[1]
Pharmacologic study revealed it to be potent and of low acute and subacute
toxicity, well tolerated by most mucous membranes and of a low sensitizing index
in humans.[1] Like other local anesthetics, pramocaine decreases the
permeability of neuronal membranes to sodium ions, blocking both initiation and
conduction of nerve impulses. Depolarization and repolarization of excitable
neural membranes is thus inhibited, leading to numbness.
Use[edit]
Topical anesthetics are used to relieve pain
and itching caused by conditions such as sunburn or other minor burns, insect
bites or stings, poison ivy, poison oak, poison sumac, and minor cuts and
scratches.[2] The popular itch creams Gold Bond and some forms of calamine
lotion use pramocaine hydrochloride to numb sensitive skin, as does the pain
relief variant of Neosporin and some formulations of Sarna. The hydrochloride
salt form of pramocaine is water-soluble.
Pramocaine and dibucaine are also common
ingredients in over the counter hemorrhoid preparations.
Pravastatin Sodium
普伐他汀鈉
https://en.wikipedia.org/wiki/Pravastatin
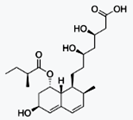
Pravastatin (marketed as Pravachol or
Selektine) is a member of the drug class of statins, used in combination with
diet, exercise, and weight loss for lowering cholesterol and preventing
cardiovascular disease.
Medical uses[edit]
Pravastatin is primarily used for the
treatment of dyslipidemia and the prevention of cardiovascular disease.[2] It is
recommended to be used only after other measures, such as diet, exercise, and
weight reduction, have not improved cholesterol levels.[2]
Mechanism of action[edit]
Pravastatin acts as a lipoprotein-lowering
drug through two pathways. In the major pathway, pravastatin inhibits the
function of hydroxymethylglutaryl-CoA (HMG-CoA) reductase. As a reversible
competitive inhibitor, pravastatin sterically hinders the action of HMG-CoA
reductase by occupying the active site of the enzyme. Taking place primarily in
the liver, this enzyme is responsible for the conversion of HMG-CoA to
mevalonate in the rate-limiting step of the biosynthetic pathway for
cholesterol. Pravastatin also inhibits the synthesis of very-low-density
lipoproteins, which are the precursor to low-density lipoproteins (LDL). These
reductions increase the number of cellular LDL receptors, thus LDL uptake
increases, removing it from the bloodstream.[7] Overall, the result is a
reduction in circulating cholesterol and LDL. A minor reduction in triglycerides
and an increase in high-density lipoproteins (HDL) are common.
Prazosin Hydrochloride
哌唑嗪鹽酸鹽
https://en.wikipedia.org/wiki/Prazosin
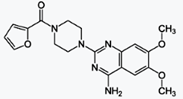
Prazosin, trade names Minipress, Vasoflex,
Lentopres and Hypovase, is a sympatholytic drug used to treat high blood
pressure, anxiety, and posttraumatic stress disorder (PTSD).[2][3] It is an
α1-blocker which acts as an inverse agonist at alpha-1 adrenergic receptors.[4]
These receptors are found on vascular smooth muscle, where they are responsible
for the vasoconstrictive action of norepinephrine.[3] They are also found
throughout the central nervous system.[5] As of 2013, prazosin is off-patent in
the US, and the FDA has approved at least one generic manufacturer.
Probucol
普羅布考
https://en.wikipedia.org/wiki/Probucol
Probucol is an anti-hyperlipidemic drug[1]
initially developed in the treatment of coronary artery disease.
However, clinical trials were stopped after
it was found that it may lower HDL in patients with a previous history of heart
disease.
Probucol was initially developed in the
1970s by a chemical company to maximize airplane tire longevity. Probucol is
associated with QT interval prolongation.
Mechanism[edit]
Probucol lowers the level of cholesterol in
the bloodstream by increasing the rate of LDL catabolism. Additionally, probucol
may inhibit cholesterol synthesis and delay cholesterol absorption.[2] Probucol
is a powerful antioxidant which inhibits the oxidation of cholesterol in LDLs;
this slows the formation of foam cells, which contribute to atherosclerotic
plaques.
It is believed to act at ABCA1.[3]
It also lowers levels of HDL.[4]https://en.wikipedia.org/wiki/ATP-binding_cassette_transporter
ATP-binding cassette transporters (ABC
transporters) are members of a transport system superfamily that is one of the
largest and is possibly one of the oldest families with representatives in all
extant phyla from prokaryotes to humans.[1][2] ABC transporters often consist of
multiple subunits, one or two of which are transmembrane proteins and one or two
of which are membrane-associated ATPases. The ATPase subunits that utilize the
energy of adenosine triphosphate (ATP) binding and hydrolysis to energize the
translocation of various substrates across membranes, either for uptake or for
export of the substrate. …..
Procainamide Hydrochloride
普魯卡因
https://en.wikipedia.org/wiki/Procainamide
Procainamide is a medication of the
antiarrhythmic class used for the treatment of cardiac arrhythmias. It is
classified by the Vaughan Williams classification system as class Ia.
Mechanism of action[edit]
Procainamide works as an anti-arrhythmic
agent and is used to treat cardiac arrhythmia. It induces rapid block of the
batrachotoxin (BTX)-activatedsodium channels of the heart muscle and acts as
antagonist to long gating closures. The block is voltage dependent and can occur
from both sides; either from the intracellular or the extracellular side.
Blocking from the extracellular side is weaker than from the intracellular side
because it occurs via the hydrophobic pathway. Procainamide is present in
charged form and probably requires a direct hydrophobic access to the binding
site for blocking of the channel. Furthermore, blocking of the channel shows a
decreased voltage sensitivity, which may result from the loss of voltage
dependence of the blocking rate. Due to its charged and hydrophilic form,
procainamide has its effect from the internal side, where it causes blockage of
voltage-dependent open channels. With increasing concentration of procainamide,
the frequency of long blockage becomes less without the duration of blockage
being affected. The rate of fast blocking is determined by the membrane
depolarization. Membrane depolarization leads to increased blocking and
decreased unblocking of the channels. Procainamide slows the conduction velocity
and increases the refractory period, such that the maximal rate of
depolarization is reduced.[7]
Propafenone Hydrochloride
普羅帕酮
https://en.wikipedia.org/wiki/Propafenone
Propafenone (/proʊˈpæfᵻnoʊn/
proh-PAF-i-nohn; brand name Rythmol SR or Rytmonorm) is a class 1C
anti-arrhythmic medication, which treats illnesses associated with rapid heart
beats such as atrial and ventricular arrhythmias.
Mechanism of action[edit]
Propafenone works by slowing the influx of
sodium ions into the cardiac muscle cells, causing a decrease in excitability of
the cells. Propafenone is more selective for cells with a high rate, but also
blocks normal cells more than class Ia or Ib. Propafenone differs from the
prototypical class Ic antiarrhythmic in that it has additional activity as a
beta-adrenergic blocker which can cause bradycardia and bronchospasm.
Pseudoephedrine base 假麻黃鹼
Pseudoephedrine Hydrochloride
Pseudoephedrine sulfate
https://en.wikipedia.org/wiki/Pseudoephedrine
Pseudoephedrine (/ˌsjuːdoʊ.ᵻˈfɛdrɪn/ or
/ˌsjuːdoʊˈɛfᵻdriːn/; PSE) is a sympathomimetic drug of the phenethylamine and
amphetamine chemical classes. It may be used as a nasal/sinus decongestant, as a
stimulant, or as a wakefulness-promoting agent.[2]
The salts pseudoephedrine hydrochloride and
pseudoephedrine sulfate are found in many over-the-counter preparations, either
as a single ingredient or (more commonly) in combination with antihistamines,
guaifenesin, dextromethorphan, and/or paracetamol (acetaminophen) or an NSAID
(such as aspirin or ibuprofen).
Mechanism of action[edit]
Pseudoephedrine is a sympathomimetic amine.
Its principal mechanism of action relies on its direct action on the adrenergic
receptor system.[8][9] The vasoconstriction that pseudoephedrine produces is
believed to be principally an α-adrenergic receptor response.[10]
Pseudoephedrine acts on α- and β2-adrenergic
receptors, to cause vasoconstriction and relaxation of smooth muscle in the
bronchi, respectively.[8][9] α-adrenergic receptors are located on the muscles
lining the walls of blood vessels. When these receptors are activated, the
muscles contract, causing the blood vessels to constrict (vasoconstriction). The
constricted blood vessels now allow less fluid to leave the blood vessels and
enter the nose, throat and sinus linings, which results in decreased
inflammation of nasal membranes, as well as decreased mucus production. Thus, by
constriction of blood vessels, mainly those located in the nasal passages,
pseudoephedrine causes a decrease in the symptoms of nasal congestion.
Activation of β2-adrenergic receptors produces relaxation of smooth muscle of
the bronchi,[8] causing bronchial dilation and in turn decreasing congestion
(although not fluid) and difficulty breathing.
Rabeprazole Sodium
雷貝拉唑鈉
https://en.wikipedia.org/wiki/Rabeprazole
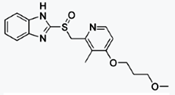
Rabeprazole /ˌræ.ˈbɛp.ræ.zɔːl/ is an
antiulcer drug in the class of proton pump inhibitors.
https://en.wikipedia.org/wiki/Proton-pump_inhibitor
It was developed by Eisai Co. and is
available worldwide under many brand names.
Indications and usage[edit]
Short-term treatment in healing and
symptomatic relief of duodenal ulcers and erosive or gastroesophageal reflux
disease (GERD); maintaining healing and reducing relapse rates of heartburn
symptoms in patients with GERD; treatment of daytime and nighttime heartburn and
other symptoms associated with GERD; long-term treatment of pathological
hypersecretory conditions, including Zollinger-Ellison syndrome and in
combination with amoxicillin and clarithromycin to eradicate Helicobacter
pylori.
•
Gastric ulcer (GU)
•
Peptic ulcer disease (PUD)
•
Maintenance of healing of erosive or ulcerative
GERD
•
Healing of erosive and ulcerative GERD
•
Healing of duodenal ulcers.
•
Treatment of symptomatic GERD
•
Treatment of pathological hypersecretory
conditions (Zollinger-Ellison syndrome)
•
Helicobacter pylori eradication to reduce risk of
duodenal ulcer recurrence
Rapamycin
雷帕黴素
https://en.wikipedia.org/wiki/Sirolimus
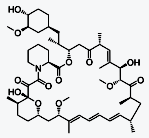
Sirolimus (INN/USAN), also known as
rapamycin, is a macrolide compound that is used to coat coronary stents, prevent
organ transplant rejection and to treat a rare lung disease called
lymphangioleiomyomatosis.[4][5][6] It has immunosuppressant functions in humans
and is especially useful in preventing the rejection of kidney transplants. It
inhibits activation of T cells and B cells by reducing the production of
interleukin-2 (IL-2).
It is produced by the bacterium Streptomyces
hygroscopicus and was isolated for the first time in 1972 by Suren Sehgal and
colleagues from samples of Streptomyces hygroscopicus found on Easter
Island.[7][8] The compound was originally named rapamycin after the native name
of the island, Rapa Nui.[5] Sirolimus was initially developed as an antifungal
agent. However, this use was abandoned when it was discovered to have potent
immunosuppressive and antiproliferative properties due to its ability to inhibit
mTOR. It was approved by the US Food and Drug Administration in September 1999
and is marketed under the trade name Rapamune by Pfizer (formerly by Wyeth).
mTOR inhibitors
https://en.wikipedia.org/wiki/MTOR_inhibitors
Rasagiline Mesylate
甲磺酸雷沙吉蘭
https://en.wikipedia.org/wiki/Rasagiline
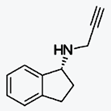
Rasagiline (Azilect, TVP-1012,
N-propargyl-1(R)-aminoindan[1]) is an irreversible inhibitor of monoamine
oxidase-B[2] used as a monotherapy to treat symptoms in early Parkinson's
disease or as an adjunct therapy in more advanced cases.[3]
The racemic form of the drug was invented by
Aspro Nicholas in the early 1979s. Moussa B.H. Youdim identified it as a
potential drug for Parkinson's disease, and working with collaborators at
Technion – Israel Institute of Technology in Israel and the drug company, Teva
Pharmaceutical, identified the R-isomer as the active form of the drug.[4] Teva
brought it to market in partnership with Lundbeck in Europe and Eisai in the US
and elsewhere. It was approved in Europe in 2005 and in the US in 2006.
Mechanism of Action[edit]
Parkinson's disease is characterized by the
death of cells that produce dopamine, a neurotransmitter. An enzyme called
monoamine oxidase (MAO) breaks down neurotransmitters. MAO has two forms, MAO-A
and MAO-B. MAO-B breaks down dopamine. Rasagiline prevents the breakdown of
dopamine by irreversibly binding to MAO-B. Dopamine is therefore more available,
somewhat compensating for the diminished quantities made in the brains of people
with Parkinsons.[5]
Selegiline was the first selective MAO-B
inhibitor. It is partly metabolized to levomethamphetamine (l-methamphetamine),
one of the two enantiomers of methamphetamine, in vivo.[9][10] While these
metabolites may contribute to selegiline's ability to inhibit reuptake of the
neurotransmitters dopamine and norepinephrine, they have also been associated
with orthostatic hypotension and hallucinations in some people.[10][11][12]
Rasagiline metabolizes into 1(R)-aminoindan which has no amphetamine-like
characteristics[13] and has neuroprotective properties in cells and in animal
models.[14]
It is selective for MAO type B over type A
by a factor of fourteen.[15]
Regadenoson
瑞加德松
https://en.wikipedia.org/wiki/Regadenoson
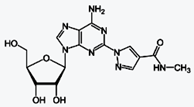
Regadenoson (CVT-3146, Lexiscan) is an A2A
adenosine receptor agonist that is a coronary vasodilator that is commonly used
in pharmacologic stress testing. It produces hyperemia quickly and maintains it
for a duration that is useful for radionuclide myocardial perfusion imaging.[1]
The selective nature of the drug makes it preferable to other stress agents such
as adenosine, which are less selective and therefore cause more side-effects.
Regadenoson was approved by the United
States Food and Drug Administration on April 10, 2008 and is marketed by
Astellas Pharma under the tradename Lexiscan.[2] It is approved for use in the
European Union and under the name of Rapiscan. It is currently being marketed by
GE Healthcare and is being sold in both the United Kingdom and Germany.
Regadenoson has a 2 to 3 minute biological
half-life, as compared with adenosine's 10-second half-life. As a result,
regadenoson stress protocols use a single bolus, instead of a 4-6 minute
continuous infusion, which was needed with adenosine. Another difference is that
adenosine infusion is weight based (140mcg/kg/minute), while with regadenoson, a
0.4mg/5mL preloaded syringe dose is standard for all weights. Regadenoson stress
tests are not affected by the presence of beta blockers, as regadenoson
vasodilates via the adenosine pathway without stimulating beta adrenergic
receptors.[citation needed]
One side effect of regadenoson is that it
can temporarily disrupt the integrity of the blood-brain barrier by inhibiting
P-glycoprotein function.[3]
Riluzole
利魯唑
https://en.wikipedia.org/wiki/Riluzole#Mechanism_of_Action
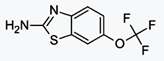
Riluzole (Rilutek, Teglutik) is a drug used
to treat amyotrophic lateral sclerosis. These are marketed by Sanofi
Pharmaceuticals and Martindale Pharma respectively. Riluzole delays the onset of
ventilator-dependence or tracheostomy in selected patients and may increase
survival by approximately two to three months.[2]
Mechanism of Action[edit]
Riluzole preferentially blocks TTX-sensitive
sodium channels, which are associated with damaged neurons.[15][16] Riluzole has
also been reported to directly inhibit the kainate and NMDA receptors.[17]
However, the action of riluzole on glutamate receptors has been controversial,
as no binding of the drug to any known sites has been shown for them.[18][19] In
addition, as its antiglutamatergic action is still detectable in the presence of
sodium channel blockers, it is also uncertain whether or not it acts via this
way. Rather, its ability to stimulate glutamate uptake seems to mediate many of
its effects.[20][21] In addition to its role in accelerating glutamate clearance
from the synapse, Riluzole may also prevent glutamate release from presynaptic
terminals.[22] These effects combined could significantly reduce glutamate
signaling and cause indirect antagonism without acting at glutamate receptors
themselves.
Rizatriptan Benzoate
利扎曲坦苯甲酸酯
https://en.wikipedia.org/wiki/Rizatriptan
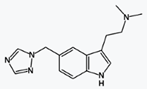
Rizatriptan (trade name Maxalt) is a 5-HT1
receptor agonist of the triptan class of drugs developed by Merck & Co. for the
treatment of migraine headaches.[1] It is available in strengths of 5 and 10 mg
as tablets and orally disintegrating tablets (Maxalt-MLT).
Maxalt obtained approval by the United
States Food and Drug Administration (FDA) on June 29, 1998. It is a
second-generation triptan.
Mechanism of action[edit]
Main article: Serotonin receptor agonist
Rizatriptan acts as an agonist at serotonin
5-HT1B and 5-HT1D receptors.[2] Like the other triptans sumatriptan and
zolmitriptan, rizatriptan induces vasoconstriction—possibly by inhibiting the
release of calcitonin gene-related peptide from sensory neurons in the
trigeminal nerve.[2]
Sebacoyl Dinalbuphine Ester
Sebacoyl Dinalbuphine Ester : a prodrug of
nalbuphine
https://en.wikipedia.org/wiki/Nalbuphine
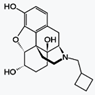
Medical uses[edit]
Nalbuphine is indicated for the relief of
moderate to severe pain. It can also be used as a supplement to balanced
anesthesia, for preoperative and postoperative analgesia, and for obstetrical
analgesia during labor and delivery.
Nalbuphine is a semi-synthetic opioid used
commercially as an analgesic under a variety of trade names, including Nubain
and Manfine.
Clinical pharmacology[edit]
Nalbuphine is a semi-synthetic opioid
agonist-antagonist analgesic of the phenanthrene series. It is chemically
related to the widely used opioid antagonists, naloxone and naltrexone, and the
potent opioid analgesic, oxymorphone.
Semi-synthetic Paclitaxel
半合成紫杉醇
https://en.wikipedia.org/wiki/Paclitaxel
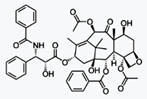
Paclitaxel (PTX), sold under the brand name
Taxol among others, is a chemotherapy medication used to treat a number of types
of cancer. This includes ovarian cancer, breast cancer, lung cancer, Kaposi
sarcoma, cervical cancer, and pancreatic cancer. It is given by injection into a
vein.[2] There is also an albumin bound formulation.[2]
Common side effects include hair loss, bone
marrow suppression, numbness, allergic reactions, muscle pains, and diarrhea.
Other serious side effects include heart problems, increased risk of infection,
and lung inflammation. Use during pregnancy may result in harm to the baby.[2]
Paclitaxel is in the taxane family of medications.[3] It works by interference
with the normal function of microtubules during cell division.[2]
Paclitaxel was first isolated in 1971 from
the Pacific yew and approved for medical use in 1993.[4][5] It is on the World
Health Organization's List of Essential Medicines, the most important medication
needed in a basic health system.[6] The wholesale cost in the developing world
is about 7.06 to 13.48 USD per 100 mg vial.[7] This amount in the United Kingdom
costs the NHS about 66.85 pounds.[8] It is now manufactured by cell culture.[5]Mechanism of action[edit]
Paclitaxel is one of several cytoskeletal
drugs that target tubulin. Paclitaxel-treated cells have defects in mitotic
spindle assembly, chromosome segregation, and cell division. Unlike other
tubulin-targeting drugs such as colchicine that inhibit microtubule assembly,
paclitaxel stabilizes the microtubule polymer and protects it from disassembly.
Chromosomes are thus unable to achieve a metaphase spindle configuration. This
blocks the progression of mitosis and prolonged activation of the mitotic
checkpoint triggers apoptosis or reversion to the G-phase of the cell cycle
without cell division.[18][19]
Sevelamer Carbonate
碳酸司维拉姆
Sevelamer Hydrochloride
https://en.wikipedia.org/wiki/Sevelamer
Sevelamer (rINN) (/sɛˈvɛləmər/ or
/sɛˈvɛləmɪər/) is a phosphate binding drug used to treat hyperphosphatemia in
patients with chronic kidney disease. When taken with meals, it binds to dietary
phosphate and prevents its absorption. Sevelamer was invented and developed by
GelTex Pharmaceuticals. Sevelamer is currently marketed by Sanofi under the
trade names Renagel (sevelamer hydrochloride) and Renvela (sevelamer carbonate).
Chemistry and pharmacology[edit]
Sevelamer consists of polyallylamine that is
crosslinked with epichlorohydrin.[1] The marketed form sevelamer hydrochloride
is a partial hydrochloride salt being present as approximately 40%
aminehydrochloride and 60% sevelamer base. The amine groups of sevelamer become
partially protonated in the intestine and interact with phosphate ions through
ionic and hydrogen bonding.
Medical uses[edit]
Sevelamer is used in the management of
hyperphosphatemia in adult patients with stage 4 and 5 chronic kidney disease on
hemodialysis. Its efficacy at lowering phosphate levels is similar to that of
calcium acetate, but without the accompanying risk of hypercalcemia.
Sodium Starch Glycolate
澱粉乙醇酸鈉
https://www.drugs.com/inactive/sodium-starch-glycolate-type-a-potato-412.html
Sodium starch glycolate type A potato is the
sodium salt of carboxymethyl ether of starch from potato origin. Starch
glycolates are also of rice, wheat or corn origin. It is a white to off-white,
tasteless, odorless, relatively free-flowing powder.
Sodium starch glycolate is used as a
pharmaceutical grade dissolution excipient for tablets and capsules. Sodium
starch glycolate absorbs water rapidly, resulting in swelling which leads to
rapid disintegration of tablets and granules. It is used as a disintegrant, a
suspending agent and as a gelling agent. Without a disintegrant, tablets may not
dissolve appropriately and may effect the amount of active ingredient absorbed,
thereby decreasing effectiveness.[1] [2]
Sodium Stearyl Fumarate
硬脂酰富馬酸鈉
https://www.drugs.com/inactive/sodium-stearyl-fumarate-320.html
Sodium stearyl fumarate is a water-soluble
lubricant used in the pharmaceutical industry for compressing tablets
("tableting").
Sodium stearyl fumarate is an inert,
hydrophilic, tablet lubricant, useful in situations where other lubricating
agents (i.e., magnesium stearate) fail to provide tablets of adequate stability,
hardness, content uniformity, disintegration and dissolution rate.[1][2]
Sodium Valproate
丙戊酸鈉
https://en.wikipedia.org/wiki/Valproate
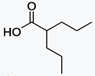
Valproate (VPA), and its valproic acid,
sodium valproate, and divalproex sodium forms, are medications primarily used to
treat epilepsy and bipolar disorder and to prevent migraine headaches.[2] It is
useful for the prevention of seizures in those with absence seizures, partial
seizures, and generalized seizures. It can be given intravenously or by mouth.
Long and short acting formulations exist.[2]
Common side effects include nausea,
vomiting, sleepiness, and a dry mouth. Serious side effects can include liver
problems and regular monitoring of liver function tests is therefore
recommended. Other serious risks include pancreatitis and an increased suicide
risk. It is known to cause serious abnormalities in the baby if taken during
pregnancy. Because of this it is not typically recommended in women of
childbearing age who have migraines. It is unclear how valproate works.[2]
Mechanism of action[edit]
Although the mechanism of action of
valproate is not fully understood,[37] it has recently been shown to protect
against a seizure-induced reduction in phosphatidylinositol
(3,4,5)-trisphosphate (PIP3) as a potential therapeutic mechanism.[49] In
addition, its anticonvulsant effect has been attributed to the blockade of
voltage-dependent sodium channels and increased brain levels of
gamma-aminobutyric acid (GABA).[37] The GABAergic effect is also believed to
contribute towards the anti-manic properties of valproate.[37] In animals,
sodium valproate raises cerebral and cerebellar levels of the inhibitory
synaptic neurotransmitter, GABA, possibly by inhibiting GABA degradative
enzymes, such as GABA transaminase, succinate-semialdehyde dehydrogenase and by
inhibiting the re-uptake of GABA by neuronal cells.[37]
It also has histone deacetylase-inhibiting
effects. The inhibition of histone deacetylase, by promoting more
transcriptionally active chromatin structures, likely presents the epigenetic
mechanism for regulation of many of the neuroprotective effects attributed to
valproic acid. Intermediate molecules mediating these effects include VEGF,
BDNF, and GDNF.[50][51]
Valproic acid has been found to be an
antagonist of the androgen and progesterone receptors, and hence as a
non-steroidal antiandrogen and antiprogestogen, at concentrations much lower
than therapeutic serum levels.[52] In addition, the drug has been identified as
a potent aromatase inhibitor, and suppresses estrogen concentrations.[53] These
actions are likely to be involved in the reproductive endocrine disturbances
seen with valproic acid treatment.[52][53]
Sulcaine
蘇爾卡因
https://pubchem.ncbi.nlm.nih.gov/compound/3300
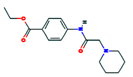
Tacrolimus
他克莫司
https://en.wikipedia.org/wiki/Tacrolimus
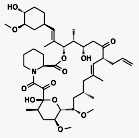
Tacrolimus (also FK-506 or fujimycin, trade
names Prograf, Advagraf, Protopic) is an immunosuppressive drug used mainly
after allogeneic organ transplant to lower the risk of organ rejection. It
achieves this by inhibiting the production of interleukin-2, a molecule that
promotes the development and proliferation of T cells, which are vital to the
body's learned (or adaptive) immune response. Tacrolimus is also used in the
treatment of other T cell-mediated diseases such as eczema (for which it is
applied to the skin in a medicated ointment), severe refractory uveitis after
bone marrow transplants, exacerbations of minimal change disease, Kimura's
disease, and the skin condition vitiligo.
Chemically it is a 23-membered macrolide
lactone that was first discovered in 1987 from the fermentation broth of a
Japanese soil sample that contained the bacterium Streptomyces tsukubaensis.
Mechanism of action[edit]
Tacrolimus is a macrolide calcineurin
inhibitor. In T-cells, activation of the T-cell receptor normally increases
intracellular calcium, which acts via calmodulin to activate calcineurin.
Calcineurin then dephosphorylates the transcription factor nuclear factor of
activated T-cells (NF-AT), which moves to the nucleus of the T-cell and
increases the activity of genes coding for IL-2 and related cytokines.
Tacrolimus prevents the dephosphorylation of NF-AT.[16]
In detail, tacrolimus reduces peptidylprolyl
isomerase activity by binding to the immunophilin FKBP12 (FK506 binding
protein), creating a new complex. This FKBP12–FK506 complex interacts with and
inhibits calcineurin, thus inhibiting both T-lymphocyte signal transduction and
IL-2 transcription.[17] Although this activity is similar to that of
ciclosporin, the incidence of acute rejection is reduced by tacrolimus use over
ciclosporin use.[1] Although short-term immunosuppression concerning patient and
graft survival is found to be similar between the two drugs, tacrolimus results
in a more favorable lipid profile, and this may have important long-term
implications given the prognostic influence of rejection on graft survival.[18]
Tafluprost
他氟前列腺素
https://en.wikipedia.org/wiki/Tafluprost
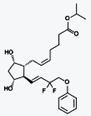
Tafluprost (trade names Taflotan by Santen
Pharmaceutical and Zioptan by Merck in the US) is a prostaglandin analogue. It
is used topically (as eye drops) to control the progression of open-angle
glaucoma and in the management of ocular hypertension, alone or in combination
with other medication. It reduces intraocular pressure by increasing the outflow
of aqueous fluid from the eyes.[1][2]
Mechanism of action[edit]
Tafluprost is a prodrug of the active
substance, tafluprost acid, a structural and functional analogue of
prostaglandin F2α (PGF2α). Tafluprost acid is a selective agonist at the
prostaglandin F receptor, increasing outflow of aqueous fluid from the eyes and
thus lowering intraocular pressure.[2][3]
Other PGF2α analogues with the same
mechanism include latanoprost and travoprost.[2]
Taltirelin
他替瑞林
https://en.wikipedia.org/wiki/Taltirelin
Taltirelin (marketed under the tradename
Ceredist) is a thyrotropin-releasing hormone (TRH) analog, which mimics the
physiological actions of TRH, but with a much longer half-life and duration of
effects,[1] and little development of tolerance following prolonged dosing.[2]
It has nootropic,[3]neuroprotective[4] and analgesic effects.[5]
Taltirelin is primarily being researched for
the treatment of spinocerebellar ataxia; limited research has also been carried
out with regard to other neurodegenerative disorders, e.g., spinal muscular
atrophy.[6][7][8]
https://en.wikipedia.org/wiki/Thyrotropin-releasing_hormone
Thyrotropin-releasing hormone (TRH), also
called thyrotropin-releasing factor (TRF) or thyroliberin, is a releasing
hormone, produced by the hypothalamus, that stimulates the release of
thyrotropin (thyroid-stimulating hormone or TSH) and prolactin from the anterior
pituitary. It is a tropic, tripeptidal hormone.
TRH has been used clinically for the
treatment of spinocerebellar degeneration and disturbance of consciousness in
humans.[1] Its pharmaceutical form is called protirelin (INN) (/proʊˈtaɪrᵻlᵻn/).
Tamsulosin HCl
坦洛新
https://en.wikipedia.org/wiki/Tamsulosin
Tamsulosin, sold under the trade name
Flomax, is a medication used to treat symptomatic benign prostatic hyperplasia
(BPH), help with the passage of kidney stones,[2] and for urinary retention
along with other measures.
Tamsulosin, and other medications in the
class called alpha blockers, work by relaxing bladder neck muscles and muscle
fibers in the prostate itself and make it easier to urinate.[3] It is an α1a
adrenergic receptor antagonist.
Tamsulosin was developed by Yamanouchi
Pharmaceuticals (now part of Astellas Pharma) and was first marketed in 1996.
The U.S. patent expired in October 2009.[4] The U.S. Food and Drug
Administration (FDA) approved generics in March 2010.[5]
Mechanism[edit]
Main article: Alpha blocker
Tamsulosin is a selective α1 receptor
antagonist that has preferential selectivity for the α1A receptor in the
prostate versus the α1B receptor in the blood vessels.[19]
When alpha 1 receptors in the bladder neck
and the prostate are blocked, this causes a relaxation in smooth muscle and
therefore less resistance to urinary flow. Due to this, the pain associated with
BPH can be reduced.
Selective action of tamsulosin in alpha 1A/D
receptors is controversial and over three quarters of tamsulosin registered
human studies are unpublished.[20]
Tegafur
https://en.wikipedia.org/wiki/Tegafur
Tegafur (INN, BAN, USAN) is a
chemotherapeutic fluorouracil prodrug used in the treatment of cancers. It is a
component of the combination drug tegafur/uracil. When metabolised, it becomes
5-fluorouracil.[1]
Mechanism of action[edit]
It is a prodrug to fluorouracil which is a
thymidylate synthase inhibitor.[2]https://en.wikipedia.org/wiki/Thymidylate_synthase
Thymidylate synthetase (EC 2.1.1.45)[4] is
an enzyme that catalyzes the conversion of deoxyuridine monophosphate (dUMP) to
deoxythymidine monophosphate (dTMP). Thymidine is one of the nucleotides in DNA.
With inhibition of TS, an imbalance of deoxynucleotides and increased levels of
dUMP arise. Both cause DNA damage.[5][6]
Telavancin HCl
特拉萬星
https://en.wikipedia.org/wiki/Telavancin
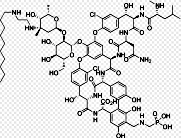
Telavancin (trade name Vibativ) is a
bactericidal lipoglycopeptide for use in MRSA or other Gram-positive infections.
Telavancin is a semi-synthetic derivative of vancomycin.[1][2]
The FDA approved the drug in September 2009
for complicated skin and skin structure infections (cSSSI),[3] and in June 2013
for hospital-acquired and ventilator-associated bacterial pneumonia caused by
Staphylococcus aureus.[4]
Mechanism of action[edit]
Like vancomycin, telavancin inhibits
bacterial cell wall synthesis by binding to the D-Ala-D-Ala terminus of the
peptidoglycan in the growing cell wall (see Pharmacology and chemistry of
vancomycin). In addition, it disrupts bacterial membranes by
depolarization.[2][9]
Temozolomide
替莫唑胺
https://en.wikipedia.org/wiki/Temozolomide
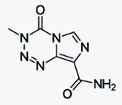
Temozolomide (TMZ; brand names Temodar and
Temodal and Temcad) is an oral chemotherapy drug. It is an alkylating agent used
as a treatment of some brain cancers; as a second-line treatment for astrocytoma
and a first-line treatment for glioblastoma multiforme.[1][2]
Mechanism of action[edit]
The therapeutic benefit of temozolomide
depends on its ability to alkylate/methylate DNA, which most often occurs at the
N-7 or O-6 positions of guanine residues. This methylation damages the DNA and
triggers the death of tumor cells. However, some tumor cells are able to repair
this type of DNA damage, and therefore diminish the therapeutic efficacy of
temozolomide, by expressing a protein O6-alkylguanine DNA alkyltransferase (AGT)
encoded in humans by the O-6-methylguanine-DNA methyltransferase (MGMT) gene.[4]
In some tumors, epigenetic silencing of the MGMT gene prevents the synthesis of
this enzyme, and as a consequence such tumors are more sensitive to killing by
temozolomide.[5] Conversely, the presence of AGT protein in brain tumors
predicts poor response to temozolomide and these patients receive little benefit
from chemotherapy with temozolomide.[6]
Thiamine Disulfide
硫胺二硫化物
https://en.wikipedia.org/wiki/Thiamine
https://www.drugs.com/ingredient/thiamine.html
Thiamine, also known as vitamin B1, is a
vitamin found in food and used as a dietary supplement.[2]As a supplement it is
used to treat and prevent thiamine deficiency and disorders that result from it
including beriberi and Korsakoff's syndrome. Other uses include maple syrup
urine disease and Leigh's disease. It is taken by mouth or by injection.[1]
Side effects are generally few. Allergic
reactions including anaphylaxis may occur. Thiamine is in the B complex family.
It is needed for the metabolism of carbohydrates.[1] As people are unable to
make it, thiamine is an essential nutrient. Food sources include whole grains,
meat, and fish.[2]
Thiamine was discovered in 1897, isolated in
1926, and first made in 1936.[3] It is on the World Health Organization's List
of Essential Medicines, the most important medication needed in a basic health
system.[4]
https://en.wikipedia.org/wiki/Vitamin
A vitamin is an organic compound and a vital
nutrient that an organism requires in limited amounts. An organic chemical
compound (or related set of compounds) is called a vitamin when the organism
cannot synthesize the compound in sufficient quantities, and it must be obtained
through the diet; thus, the term "vitamin" is conditional upon the circumstances
and the particular organism.
Thiopental
硫噴妥鈉
https://en.wikipedia.org/wiki/Sodium_thiopental
Sodium thiopental, also known as Sodium
Pentothal (a trademark of Abbott Laboratories, not to be confused with
pentobarbital), thiopental, thiopentone, or Trapanal (also a trademark), is a
rapid-onset short-acting barbiturate general anesthetic that is an analogue of
thiobarbital. Sodium thiopental was a core medicine in the World Health
Organization's "Essential Drugs List", which is a list of minimum medical needs
for a basic healthcare system, but was supplanted by propofol.[3] It was
previously the first of three drugs administered during most lethal injections
in the United States, but the U.S. manufacturer Hospira stopped manufacturing
the drug and the EU banned the export of the drug for this purpose.[4]
Mechanism of action[edit]
Main article: Barbiturate
Sodium thiopental is a member of the
barbiturate class of drugs, which are relatively non-selective compounds that
bind to an entire superfamily of ligand-gated ion channels, of which the GABAA
receptor channel is one of several representatives. This superfamily of ion
channels includes the neuronal nAChR channel, the 5HT3R channel, the GlyR
channel and others. Surprisingly, while GABAA receptor currents are increased by
barbiturates (and other general anesthetics), ligand-gated ion channels that are
predominantly permeable for cationic ions are blocked by these compounds. For
example, neuronal nAChR channels are blocked by clinically relevant anesthetic
concentrations of both sodium thiopental and pentobarbital.[23] Such findings
implicate (non-GABA-ergic) ligand-gated ion channels, e.g. the neuronal nAChR
channel, in mediating some of the (side) effects of barbiturates.[24] The GABAA
receptor is an inhibitory channel that decreases neuronal activity, and
barbiturates enhance the inhibitory action of the GABAA receptor.[25]
Timolol Maleate
馬來酸噻嗎洛爾
https://en.wikipedia.org/wiki/Timolol
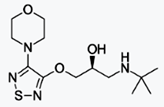
Timolol is a medication used either by mouth
or as eye drops.[2][3] As eye drops it is used to treat increased pressure
inside the eye such as in ocular hypertension and glaucoma.[2] By mouth it is
used for high blood pressure, chest pain due to not enough blood flow to the
heart, to prevent further complications after a heart attack, and to prevent
migraines.[3]
Common side effects with the drops is
irritation of the eye.[2] Common side effects by mouth include feeling tired,
slow heart beat, itchiness, and shortness of breath.[3] Other side effects
include masking the symptoms of low blood sugar in those with diabetes. Use is
not recommended in those with asthma, heart failure, or COPD.[2] It is unclear
if use during pregnancy is safe for the baby.[4] Timolol is in the non-selective
Beta blocker family of medication.[2]
Tipepidine Hibenzate
阿斯维林;提培匹定;
双噻甲哌啶;海苯酸替培啶;羟苯酰苯酸替培啶;双噻甲哌啶 2-(4-羟基苯甲酰)苯甲酸盐;3-[二(噻吩-2-基)亚甲基]-1-甲基哌啶 2-(4-羟基苯甲酰)苯甲酸盐 (1:1)http://www.chemicalbook.com/chemicalproductproperty_cn_cb1299803.htm
https://en.wikipedia.org/wiki/Tipepidine
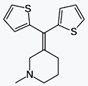
Tipepidine (INN) (brand names Asverin,
Antupex, Asvelik, Asvex, Bitiodin, Cofdenin A, Hustel, Nodal, Sotal), also known
as tipepidine hibenzate (JAN), is a synthetic, non-opioid antitussive and
expectorant of the thiambutene class.[1][2] It acts as an inhibitor of G
protein-coupled inwardly-rectifying potassium channels (GIRKs).[3] The drug was
discovered in the 1950s,[4] and was developed in Japan in 1959.[5] It is used as
the hibenzate and citrate salts.[1][5]
The usual dose is 20 mg every 4–6
hours.[citation needed] Possible side effects of tipepidine, especially in
overdose, may include drowsiness, vertigo, delirium, disorientation, loss of
consciousness, and confusion.[5]
Tipepidine has recently garnered interest as
a potential psychiatric drug. It is being investigated in depression,[3][6][7]
obsessive-compulsive disorder,[8] and attention-deficit hyperactivity disorder
(ADHD).[9][10] Through inhibition of GIRK channels, tipepidine increases
dopamine levels in the nucleus accumbens, but without increasing locomotor
activity or producing methamphetamine-like behavioral sensitization, and this
action appears to be at least partly responsible for its antidepressant-like
effects in rodents.[11][12]
Tolperisone HCl
托哌酮
https://en.wikipedia.org/wiki/Tolperisone
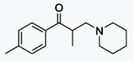
Tolperisone, a piperidine derivative, is a
centrally acting muscle relaxant. Trade names include Biocalm, Muscodol,
Mydeton, Mydocalm, Mydoflex, Myolax, Myoxan and Viveo.
Mechanism of action[edit]
Tolperisone is a centrally acting muscle
relaxant that acts at the reticular formation in the brain stem[1] by blocking
voltage-gated sodium and calcium channels.[7][8]
Topiramate
托吡酯
https://en.wikipedia.org/wiki/Topiramate
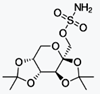
Topiramate (brand name Topamax) is an
anticonvulsant (antiepilepsy) drug. In late 2012, topiramate was approved by the
United States Food and Drug Administration (FDA) in combination with phentermine
for weight loss. The drug had previously been used off-label for this purpose.
Topiramate was originally produced by Ortho-McNeil Neurologics and Noramco,
Inc., both divisions of the Johnson & Johnson Corporation. This medication was
discovered in 1979 by Bruce E. Maryanoff and Joseph F. Gardocki during their
research work at McNeil Pharmaceutical.[1][2][3]
Topiramate came into commercial use in
1996.[4] Generic versions are available in Canada and these were approved by the
FDA in September 2006. Mylan Pharmaceuticals was recently granted final approval
for generic topiramate by the FDA for sale in the United States.[5] The last
patent for topiramate in the U.S. was for use in children and expired on
February 28, 2009.[6]
Anticonvulsant:
https://en.wikipedia.org/wiki/Anticonvulsant
Conventional antiepileptic drugs may block
sodium channels or enhance γ-aminobutyric acid (GABA) function. Several
antiepileptic drugs have multiple or uncertain mechanisms of action.[7] Next to
the voltage-gated sodium channels and components of the GABA system, their
targets include GABAA receptors, the GAT-1 GABA transporter, and GABA
transaminase.[8] Additional targets include voltage-gated calcium channels,
SV2A, and α2δ.[9][10] By blocking sodium or calcium channels, antiepileptic
drugs reduce the release of excitatory glutamate, whose release is considered to
be elevated in epilepsy, but also that of GABA.[11] This is probably a side
effect or even the actual mechanism of action for some antiepileptic drugs,
since GABA can itself, directly or indirectly, act proconvulsively.[11] Another
potential target of antiepileptic drugs is the peroxisome proliferator-activated
receptor alpha.[12][13][14][15][16][17][18] The drug class was the
5th-best-selling in the US in 2007.[19]
Topotecan HCl
托泊替康鹽酸鹽
https://en.wikipedia.org/wiki/Topotecan
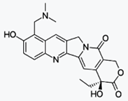
Topotecan (trade name Hycamtin) is a
chemotherapeutic agent that is a topoisomerase inhibitor. It is a synthetic,
water-soluble analog of the natural chemical compound camptothecin. It is used
in the form of its hydrochloride salt to treat ovarian cancer, lung cancer and
other cancer types.
After GlaxoSmithKline received final FDA
approval for Hycamtin Capsules on October 15, 2007, topotecan became the first
topoisomerase I inhibitor for oral use.
Mechanism of action[edit]
Topotecan is a semi-synthetic derivative of
camptothecin. Camptothecin is a natural product extracted from the bark of the
tree Camptotheca acuminata. Topoisomerase-I is a nuclear enzyme that relieves
torsional strain in DNA by opening single strand breaks.[16] Once
topoisomerase-I creates a single strand break, the DNA can rotate in front of
the advancing replication fork. In physiological environments, topotecan is in
equilibrium with its inactive carboxylate form.[17] Topotecan's active lactone
form intercalates between DNA bases in the topoisomerase-I cleavage complex.[18]
The binding of topotecan in the cleavage complex prevents topoisomerase-I from
religating the nicked DNA strand after relieving the strain.[18] This
intercalation therefore traps the topoisomerase-I in the cleavage complex bound
to the DNA.[18] When the replication-fork collides with the trapped
topoisomerase-I, DNA damage occurs.[18] The unbroken DNA strand breaks and
mammalian cells cannot efficiently repair these double strand breaks.[19] The
accumulation of trapped topoisomerase-I complexes is a known response to
apoptotic stimuli.[20] This disruption prevents DNA replication and ultimately
leads to cell death. This process leads to breaks in the DNA strand resulting in
apoptosis. Administration of topotecan down-regulates its target,
topoisomerase-I; therefore, it is dosed to maximize efficacy and minimize
related toxicity.[17] Topotecan is often given in combination with Paclitaxel as
first line treatment for extensive-stage small-cell lung cancer.[17]
Trandolapril
群多普利
https://en.wikipedia.org/wiki/Trandolapril
Trandolapril is an ACE inhibitor used to
treat high blood pressure, it may also be used to treat other conditions. It is
marketed by Abbott Laboratories under the brand name Mavik.
Trandolapril is a prodrug that is
de-esterified to trandolaprilat. It is believed to exert its antihypertensive
effect through the renin-angiotensin-aldosterone system. Trandolapril has a
half-life of about 6 hours, and trandolaprilat has a half life of about 10 h.
Trandolaprilat has about eight times the activity of its parent drug. About
one-third of trandolapril and its metabolites are excreted in the urine, and
about two-thirds of trandolapril and its metabolites are excreted in the feces.
Serum protein binding of trandolapril is about 80%.
Mode of action[edit]
Trandolapril acts by competitive inhibition
of angiotensin converting enzyme (ACE), a key enzyme in the renin-angiotensin
system which plays an important role in regulating blood pressure.
Travoprost
曲伏前列素
https://en.wikipedia.org/wiki/Travoprost
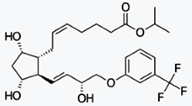
Travoprost ophthalmic solution is a topical
medication used for controlling the progression of glaucoma or ocular
hypertension, by reducing intraocular pressure. It is a synthetic prostaglandin
analog (or more specifically, an analog of prostaglandin F2α)[1][2] that works
by increasing the outflow of aqueous fluid from the eyes.[3] It is also known by
the brand names of Travatan and Travatan Z, manufactured by Alcon.
Mechanism of action[edit]
Like other analogs of prostaglandin F2α such
as tafluprost and latanoprost, travoprost is an esterprodrug of the free acid,
which acts as an agonist at the prostaglandin F receptor, increasing outflow of
aqueous fluid from the eye and thus lowering intraocular pressure.[4]
Treprostinil
曲前列素
Treprostinil Sodium
https://en.wikipedia.org/wiki/Treprostinil
Treprostinil (marketed under the trade names
Remodulin for infusion, Orenitram for oral, and Tyvaso for inhalation) is a
vasodilator that is used for the treatment of pulmonary arterial
hypertension.[1] Treprostinil is a synthetic analog of prostacyclin (PGI2).
The inhaled form of treprostinil was
approved by the FDA in July 2009 and is marketed as the trade name Tyvaso.
Mechanism of action[edit]
The major effects of treprostinil are
vasodilation of arteries in the lungs and body. Treprostinil also inhibits
platelet aggregation and smooth muscle proliferation.
Mode of action[edit]
https://en.wikipedia.org/wiki/Prostacyclin#Mode_of_action
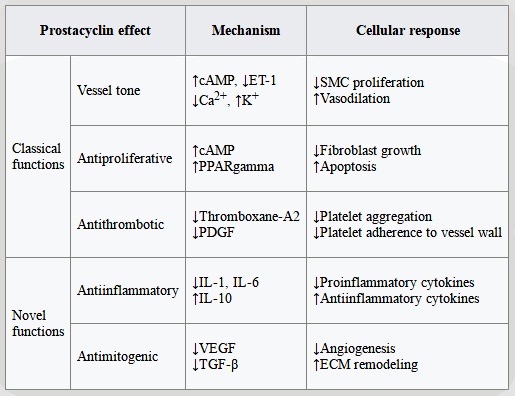
Unoprostone Isopropyl
烏諾前列酮
https://en.wikipedia.org/wiki/Unoprostone
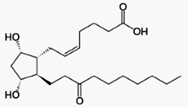
Unoprostone (INN) is a prostaglandin
analogue. Its isopropyl ester, unoprostone isopropyl, was marketed under the
trade name Rescula for the management of open-angle glaucoma and ocular
hypertension, but is now discontinued in the US.[1]
Valproic acid
丙戊酸
https://en.wikipedia.org/wiki/Valproate
As: Divalproex Sodium
Vilazodone HCl
維拉佐酮
https://en.wikipedia.org/wiki/Vilazodone
Vilazodone (United States trade name Viibryd
VEYE-brid) is a serotonergic antidepressant developed by Clinical Data for the
treatment of major depressive disorder. The chemical compound was originally
developed by Merck KGaA (Germany).[2] Vilazodone was approved by the FDA for use
in the United States to treat major depressive disorder in 2011.[3][4][5] Its
activity can be thought of as a combination of an SSRI and buspirone in some
ways.
Pharmacology[edit]
Vilazodone acts as a serotonin reuptake
inhibitor (IC50 = 2.1 nM; Ki = 0.1 nM) and 5-HT1A receptor partial agonist (IC50
= 0.2 nM; IA = ~60–70%).[6][11] It has negligible affinity for other serotonin
receptors such as 5-HT1D, 5-HT2A, and 5-HT2C.[11][12] It also exhibits
negligible inhibitory activity at the norepinephrine and dopamine transporters
(IC50 = 56 nM for NET and 37 nM for DAT).[1]
Zoledronic acid
唑來膦酸
https://en.wikipedia.org/wiki/Zoledronic_acid
Zoledronic acid (INN) or zoledronate is a
bisphosphonate drug given intravenously to treat some bone diseases. It is sold
under many trade names worldwide.[1]
Mechanism of action[edit]
Zoledronic acid slows down bone resorption,
allowing the bone-forming cells time to rebuild normal bone and allowing bone
remodeling.[2]
Medical uses[edit]
Bone complications of cancer[edit]
Zometa is used to prevent skeletal fractures
in patients with cancers such as multiple myeloma and prostate cancer, as well
as for treating osteoporosis.[3] It can also be used to treat hypercalcemia of
malignancy and can be helpful for treating pain from bone metastases.[4]
It can be administered at home rather than
in hospital. Such administration has shown safety and quality-of-life benefits
in breast cancer patients with bone metastases.[5]
Osteoporosis[edit]
Marketed as Aclasta (in Australia) or
Reclast (in the US), zoledronic acid may be given as a 5 mg infusion once per
year for treatment of osteoporosis in men and post-menopausal women at increased
risk of fracture.[medical citation needed]
In 2007, the U.S. Food and Drug
Administration (FDA) also approved Reclast for the treatment of postmenopausal
osteoporosis.[medical citation needed]
Zolmitriptan
佐米曲普坦
https://en.wikipedia.org/wiki/Zolmitriptan
Zolmitriptan is a selective serotonin
receptor agonist of the 1B and 1D subtypes. It is a triptan, used in the acute
treatment of migraine attacks with or without aura and cluster headaches.
Zolmitriptan is marketed by AstraZeneca with
the brand names Zomig, Zomigon (Argentina, Canada & Greece), AscoTop (Germany)
and Zomigoro (France). In 2008, Zomig generated nearly $154 million in sales.[1]
AstraZeneca's U.S. patent on Zomig tablets
expired on November 14, 2012, and its pediatric exclusivity extension expired on
May 14, 2013.[2] The patent in certain European countries has already expired
too, and generic drug maker Actavis released a generic version in those
countries, starting in March 2012.[3]
Zotarolimus
佐他莫司
https://en.wikipedia.org/wiki/Zotarolimus
Zotarolimus (INN, codenamed ABT-578) is an
immunosuppressant. It is a semi-synthetic derivative of rapamycin. It was
designed for use in stents with phosphorylcholine as a carrier. Coronary stents
reduce early complications and improve late clinical outcomes in patients
needing interventional cardiology.[1] The first human coronary stent
implantation was first performed in 1986 by Puel et al.[1][2] However, there are
complications associated with stent use, development of thrombosis which impedes
the efficiency of coronary stents, haemorrhagic and restenosis complications are
problems associated with stents.[1]
These complications have prompted the
development of drug-eluting stents. Stents are bound by a membrane consisting of
polymers which not only slowly release zotarolimus and its derivatives into the
surrounding tissues but also do not invoke an inflammatory response by the body.
Medtronic are using zotarolimus as the
anti-proliferative agent in the polymer coating of their Endeavor and Resolute
products.[3]
|
|