|
Global Pharmaceutical API Products
全球原料藥廠生產品項
Top Level Index
Gastrointestinal tract / Alimentary system
Antibiotics
Antidiabetic & pcos
Cardiovascular system
Central nervous system
Contraceptive agents
Dermatologicals
Opthalmic
Genito-urinary tract
Hormones
Immunology
Metabolism
Musculo - skeletal disorder
Oncology
Oropharyngeal
Respiratory system & anti- allergics
Supplementation
Surgical & Vaccines
Veterinary list
We manufacture
Index / Table of Content
Summary By Fucntion
|
Gastrointestinal tract / Alimentary system |
|
Antacids, Antireflux Agents & Antiulcerants |
Homatropine Methylbromide
https://en.wikipedia.org/wiki/Homatropine_methylbromide
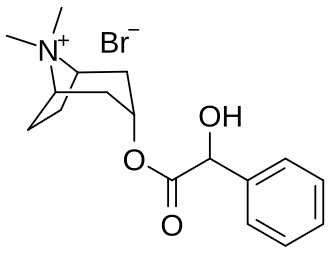
Homatropine methylbromide (INN; also known as methylhomatropine bromide) is a quaternary ammonium salt of methylhomatropine. It is a peripherally acting anticholinergic medication that inhibits muscarinic
acetylcholine receptors and thus the parasympathetic nervous system. It does not cross the blood–brain barrier. It is used to effectively relieve intestinal spasms and abdominal cramps, without producing the adverse effects of less specific anticholinergics.It is used, in addition to papaverine,
as a component of mild drugs that help "flush" the bile.
Certain preparations of drugs such as hydrocodone are mixed with a small, sub-therapeutic amount of homatropine methylbromide to discourage intentional overdose.
|
Mesalazine
https://en.wikipedia.org/wiki/Mesalazine

Mesalazine, also known as mesalamine or 5-aminosalicylic
acid (5-ASA),
is a medication used to treat inflammatory bowel disease,
including ulcerative colitis and Crohn's
disease. It
is generally used for mildly to moderately severe disease. It
is taken by mouth or rectally.The
formulations which are taken by mouth appear to be similarly effective.
It is used to treat inflammatory
bowel disease, including ulcerative
colitis and Crohn's
disease. It
is generally used for mildly to moderately severe disease. It
is taken by mouth or rectally. Thformulations
which are taken by mouth appear to be similarly effective.
|
Rebamipide
https://en.wikipedia.org/wiki/Rebamipide

Rebamipide, an amino acid derivative of 2-(1H)-quinolinone, is used for mucosal protection, healing of gastroduodenal ulcers, and treatment of gastritis.It works by enhancing mucosal defense, scavenging free radicals and
temporarily activating genes encoding cyclooxygenase-2
Studies have shown that rebamipide can fight the damaging effects of NSAIDs on the GIT mucosa and more recently, the small intestine, but not for naproxen-induced gastric damage.
|
Sucralfate
https://en.wikipedia.org/wiki/Sucralfate

Sucralfate, sold under various brand names, is
a medication used
to treat stomach ulcers, gastroesophageal
reflux disease (GERD), radiation
proctitis, and stomach inflammation and
to prevent stress ulcers.
Its usefulness in people infected by H. pylori is
limited. It is used by mouth and rectally
Sucralfate is a locally acting substance that in an acidic environment (pH < 4) reacts with hydrochloric acid in the stomach to form a cross-linking, viscous, paste-like material capable of acting as an acid
buffer for as long as 6 to 8 hours after a single dose. It also attaches to proteins on the surface of ulcers, such as albumin and fibrinogen, to form stable insoluble complexes.
These complexes serve as protective barriers at the ulcer surface, preventing further damage from acid, pepsin, and bile In addition, sucralfate prevents back diffusion of hydrogen ions, and adsorbs both pepsin and bile
acids.
It has been thought that sucralfate also stimulates the production of prostaglandin E2, epidermal growth factors (EGF), bFGF, and gastric mucus
|
Magaldrate
https://en.wikipedia.org/wiki/Magaldrate

Magaldrate (INN)
is a common antacid drug that
is used for the treatment of duodenal and gastric
ulcers, esophagitis from gastroesophageal
reflux.Magaldrate is a hydroxymagnesium aluminate complex that is converted rapidly by gastric acid into Mg(OH)2 and
Al(OH)3, which are absorbed poorly and thus provide a sustained antacid effect. |
|
Constipation & Bowel Cleansers |
Docusate Sodium
https://en.wikipedia.org/wiki/Docusate

Docusate is the common chemical and pharmaceutical name of the anion bis(2-ethylhexyl) sulfosuccinate, also commonly called dioctyl sulfosuccinate (DOSS).
Salts of this anion, especially docusate sodium, are widely used in medicine as laxatives and as stool softeners, by mouth or rectally.Docusate sodium is on the WHO
List of Essential Medicines. It is a widely available and relatively inexpensive generic medication, with more than six million prescriptions in the US in 2017.Other docusate salts with medical use include those of calcium, sodium, and potassium.
Docusate salts are also used as food additives, emulsifiers, dispersants, and wetting agents, among other uses
|
| |
Prucalopride Succinate
https://en.wikipedia.org/wiki/Prucalopride

Prucalopride, brand name Prudac,
among others, is a drug acting
as a selective, high affinity 5-HT4 receptor agonist which
targets the impaired motility associated with chronic constipation,
thus normalizing bowel movements. Prucalopride
was approved for use in Europe in 2009,in Canada in 2011 and
in Israel in 2014 but
has only been recently approved by the Food and Drug Administration for
use in the United States. The drug has also been tested for the treatment of chronic intestinal pseudo-obstruction. |
| |
Bisacodyl
https://en.wikipedia.org/wiki/Bisacodyl

Bisacodyl (INN) is an organic compound that is used as a stimulant laxative drug. It works directly on the colon to
produce a bowel movement. It is typically prescribed for relief of episodic and chronic constipation and for the management of neurogenic bowel dysfunction, as well as part of bowel preparation before medical examinations, such as for a colonoscopy.
Bisacodyl is a derivative of triphenylmethane. It was first used as a laxative in 1953 because of its structural similarity to phenolphthalein.Bisacodyl works by stimulating enteric
nerves to cause peristalsis,
i.e., colonic contractions. It is also a contact laxative; it increases fluid and salt secretion. The action of bisacodyl on the small
intestine is negligible; stimulant laxatives mainly promote evacuation of the colon.
. |
| Dissolution Of Cholestrol - Rich Gallstones |
Ursodeoxycholic Acid
https://en.wikipedia.org/wiki/Ursodeoxycholic_acid

Ursodeoxycholic acid (UDCA),
also known as ursodiol, is a secondary bile acid, produced in humans and most other
species from metabolism by intestinal bacteria. It is synthesized in the liver in some species, and was first identified in bear bile, which is the derivation of its name Ursus.
In purified form, it has been used to treat or prevent several diseases of the liver or bile
ducts.
Primary Primary bile acids are produced by the liver and
stored in the gall bladder. When secreted into the intestine, primary bile acids can be metabolized into secondary bile acids by intestinal bacteria. Primary and secondary bile acids help the body digest fats.
Ursodeoxycholic acid helps regulate cholesterol by
reducing the rate at which the intestine absorbs
cholesterol molecules while breaking up micelles containing
cholesterol. The drug reduces cholesterol absorption and is used to dissolve (cholesterol) gallstones in patients who
want an alternative to surgery.There
are multiple mechanisms involved in cholestatic liver diseases.
|
| Drug Modifying Intestinal Motility & Acid Secretions |
Hyoscine Butylbromide
https://en.wikipedia.org/wiki/Hyoscine_butylbromide

Hyoscine butylbromide, also known as scopolamine
butylbromide and sold under
the brandname Buscopan among
others,[3] is
a medication used to treat crampy abdominal pain, esophageal
spasms, renal
colic, and bladder spasms.It
is also used to improve respiratory secretions at
the end of life. Hyoscine
butylbromide can be taken by mouth, injection into a muscle, or into
a vein.
Hyoscine butylbromide is effective in treating crampy abdominal pain.Hyoscine butylbromide is effective in reducing the duration of the first stage of labour, and it is not associated with any obvious adverse outcomes in mother or neonate.It is also used during abdominal or pelvic MRI or CT scans to
improve the quality of pictures.
|
Metoclopramide
https://en.wikipedia.org/wiki/Metoclopramide

Metoclopramide is a medication used mostly for stomach and esophageal problems It is commonly used to treat and prevent nausea and vomiting, to help with emptying
of the stomach in people with delayed stomach emptying, and to help with gastroesophageal reflux disease.It is also used to treat migraine headaches
In 2012, metoclopramide was one of the top 100 most prescribed medications in the United States.It is available as a generic medication.It is on the World Health Organization's List of Essential Medicines In 2017, it was the 253rd most commonly prescribed medication in the United States, with more than one million prescriptions.
|
Mosapride citrate
https://en.wikipedia.org/wiki/Mosapride

Mosapride is
a gastroprokinetic agent that
acts as a selective 5HT4 agonist.
The major active metabolite of mosapride, known as M1, additionally acts as a 5HT3 antagonist, which
accelerates gastric emptying throughout
the whole of the gastrointestinal tract in humans, and
is used for the treatment of gastritis, gastroesophageal
reflux disease, functional
dyspepsia and irritable
bowel syndrome. It
is recommended to be taken on an empty stomach (i.e. at least one hour before food or two hours after food).
|
| H2 Blockers & Ulcer Healing Drugs |
Esomeprazole
https://en.wikipedia.org/wiki/Esomeprazole

Esomeprazole,
sold under the brand names Nexium among
others,[2] is
a medication which reduces stomach acid. It
is used to treat gastroesophageal reflux disease, peptic
ulcer disease, and Zollinger–Ellison
syndrome. Effectiveness
is similar to other proton pump inhibitors (PPIs). It
is taken by mouth or injection into a vein.
|
Lansoprazole
https://en.wikipedia.org/wiki/Lansoprazole

Lansoprazole, sold under the brand name Prevacid among
others, is a medication which reduces stomach acid. It
is used to treat peptic ulcer disease, gastroesophageal
reflux disease, and Zollinger–Ellison syndrome. Effectiveness
is similar to other proton pump inhibitors (PPIs). It
is taken by mouth.Onset is over a few hours and effects last up to a couple of daysIt is a racemic 1:1 mixture of the enantiomers dexlansoprazole and
levolansoprazole. Dexlansoprazole is an enantiomerically pure active ingredient of a commercial drug as a result of the enantiomeric shift. Lansoprazole's plasma elimination half-life (1.5 h) is not proportional to the duration of the drug's effects to the person (i.e. gastric
acid suppression).
|
Omeprazole
https://en.wikipedia.org/wiki/Omeprazole

Omeprazole, sold under the brand names Prilosec and Losec among
others, is a medication used in the treatment of gastroesophageal reflux disease (GERD), peptic
ulcer disease, and Zollinger–Ellison syndrome.It
is also used to prevent upper gastrointestinal bleeding in
people who are at high risk. Omeprazole
is a proton-pump inhibitor (PPI)
and its effectiveness is similar to other PPIs. It
can be taken by mouth or by injection into a vein.Omeprazole
contains a tricoordinated sulfinyl sulfur in a pyramidal structure and therefore can exist as either the (S)- or (R)-enantiomers.
Omeprazole is a racemate, an equal mixture of the two. In the acidic conditions of the canaliculi of parietal
cells, both enantiomers are converted to achiral products (sulfenic
acid and sulfenamide configurations) which react with a cysteine group
in H+/K+ ATPase,
thereby inhibiting the ability of the parietal cells to produce gastric acid.
|
Pantoprazole
https://en.wikipedia.org/wiki/Pantoprazole

Pantoprazole, sold under the brand name Protonix among
others, is a medication used for the treatment of stomach ulcers, short-term treatment of erosive
esophagitis due to gastroesophageal
reflux disease (GERD),
maintenance of healing of erosive esophagitis, and pathological hypersecretory conditions including Zollinger–Ellison syndrome. It
may also be used along with other medications to eliminate Helicobacter pylori. Effectiveness
is similar to other proton pump inhibitors (PPIs). It
is available by mouth and
by injection into a vein.
|
Rabeprazole sodium
https://en.wikipedia.org/wiki/Rabeprazole

Rabeprazole,
sold under the brand name Aciphex,
among others, is a medication that decreases stomach acid.It
is used to treat peptic ulcer disease, gastroesophageal
reflux disease, and excess stomach acid production such as in Zollinger–Ellison
syndrome. It
may also be used in combination with other medications to treat Helicobacter pylori. Effectiveness
is similar to other proton pump inhibitors (PPIs). It
is taken by mouth.Rabeprazole is classified as a substituted benzimidazole, like omeprazole, lansoprazole,
and pantoprazole. Rabeprazole
possess properties of both acids and bases, making it an amphotere.The acid
dissociation constant (pKa)
of the pyridine nitrogen is
about equal to 5.
|
Ranitidine
https://en.wikipedia.org/wiki/Ranitidine

Ranitidine, sold under the trade name Zantac among
others, is a medication that decreases stomach acid production. It
is commonly used in treatment of peptic ulcer disease, gastroesophageal
reflux disease, and Zollinger–Ellison syndrome. There
is also tentative evidence of benefit for hives. It
can be given by mouth, by injection
into a muscle, or by injection into a veinRanitidine
is a competitive, reversible inhibitor of the action of histamine at the histamine H2 receptors
found in gastric parietal cells. This results in decreased gastric acid secretion and gastric volume, and reduced hydrogen ion concentration.
|
| Non Specific Antidiarrhoeal Agents |
Loperamide
https://en.wikipedia.org/wiki/Loperamide

Loperamide, sold under the brand name Imodium,
among others, is a
medication used to decrease the frequency of diarrhea. It
is often used for this purpose in, inflammatory bowel disease, and short
bowel syndrome. It
is not recommended for those with blood in the stool,
mucus in the stool or fevers. The
medication is taken by mouth.
Loperamide is an opioid-receptor agonist and acts on the μ-opioid receptors in the myenteric plexus of the large intestine. It works like morphine, decreasing the activity of the myenteric plexus, which decreases the tone of the longitudinal and circular smooth
muscles of the intestinal wall. This increases the time material stays in the intestine, allowing more water to be absorbed from the fecal matter. It also decreases colonic mass movements and suppresses the gastrocolic reflex.
Loperamide's circulation in the bloodstream is limited in two ways. Efflux by P-glycoprotein in the intestinal wall reduces passage of loperamide, and the fraction of drug crossing is then further reduced through first-pass metabolism by the liver.Loperamide metabolizes into an MPTP-like compound, but is unlikely to exert neurotoxicity.
|
Racecadotril
https://en.wikipedia.org/wiki/Racecadotril

Racecadotril, also known as acetorphan,
is an antidiarrheal drug
which acts as a peripheral enkephalinase inhibitor.Unlike
other opioid medications
used to treat diarrhea, which reduce intestinal motility, racecadotril has an antisecretory effect — it reduces the secretion of water and electrolytes into
the intestine. It is
available in France (where it was first introduced in ~1990) and other European countries (including Germany, Italy, the United Kingdom, Spain, Portugal, Russia and the Czech Republic) as well as most of South America and some South East Asian countries (including China, India and Thailand), but not in the United States. It is sold under the tradename Hidrasec,
among others. Thiorphan is
the active metabolite of racecadotril, which exerts the bulk of its inhibitory actions on enkephalinases.
Enkephalins are peptides produced by the body that act on opioid receptors with preference for the δ subtype.[ Activation
of δ receptors inhibits the enzyme adenylyl cyclase, decreasing intracellular levels of the messenger molecule cAMP.
The active metabolite of racecadotril, thiorphan, inhibits enkephalinase enzymes in the intestinal epithelium with an IC50 of
6.1 nM, protecting enkephalins from being broken down by these enzymes. (Racecadotril itself is much less potent at 4500 nM. This reduces diarrhea related hypersecretion in the small intestine without influencing basal secretion. Racecadotril also has no influence on the time substances, bacteria or virus particles stay in the intestine.
|
Rifaximin
https://en.wikipedia.org/wiki/Rifaximin

Rifaximin, sold under the trade name Xifaxan among
others, is an antibiotic used
to treat traveler's diarrhea, irritable
bowel syndrome, and hepatic encephalopathy. It
has poor absorption when taken by mouth.
Rifaximin interferes with transcription by binding to the β-subunit of bacterial RNA polymerase. This results in the blockage of the translocation step that normally follows the formation of the first phosphodiester bond, which occurs in the transcription process.[ This
in turn results in a reduction of bacteria populations, including gas producing bacteria, which may reduce mucosal inflammation, epithelial dysfunction and visceral hypersensitivity. Rifaximin has broad spectrum antibacterial properties against both gram
positive and gram negative anaerobic and aerobic bacteria. As a result of bile acid solubility, its antibacterial action is limited mostly to the small intestine and less so the colon. A resetting of the
bacteria composition has also been suggested as a possible mechanism of action for relief of IBS symptoms.Additionally, rifaximin may have a direct anti-inflammatory effect on gut mucosa via modulation of the pregnane X receptor.
Other mechanisms for its therapeutic properties include inhibition of bacterial translocation across the epithelial lining of the intestine, inhibition of adherence of bacteria to the epithelial cells and a reduction in the expression of proinflammatory cytokines.
|
Trimethoprim
https://en.wikipedia.org/wiki/Trimethoprim

Trimethoprim (TMP)
is an antibiotic used
mainly in the treatment of bladder infections.Other
uses include for middle ear infections and travelers'
diarrhea With sulfamethoxazole or dapsone it
may be used for Pneumocystis pneumonia in
people with HIV/AIDS It
is taken by mouth.Trimethoprim binds to dihydrofolate reductase and inhibits the reduction of dihydrofolic
acid (DHF) to tetrahydrofolic acid (THF).THF is an essential precursor in the thymidine synthesis pathway and interference with this pathway inhibits bacterial DNA synthesis.Trimethoprim's affinity for bacterial dihydrofolate reductase is several thousand times greater than its affinity for human dihydrofolate reductase. Sulfamethoxazole inhibits dihydropteroate
synthase, an enzyme involved further upstream in the same pathway. Trimethoprim and sulfamethoxazole are commonly used in combination due to possible synergistic effects, and reduced development of resistance.This benefit has been questioned.
|
| Probiotics |
Saccharomyces Boulardii
https://en.wikipedia.org/wiki/Saccharomyces_boulardii

Saccharomyces boulardii is a tropical species of yeast first isolated from lychee and mangosteen fruit in 1923 by French scientist Henri Boulard. Although early reports described distinct taxonomic,
metabolic, and genetic properties S. boulardii is a strain of S. cerevisiae, sharing >99% genomic relatedness, giving the synonym S. cerevisiae var boulardii. A type strain is Hansen CBS 5926.
S. boulardii is sometimes used as a probiotic with the purpose of introducing beneficial microbes into the large and small intestines and conferring protection against pathogens.It grows at 37 °C (98.6 °F).In addition, the popular genome-editing tool CRISPR-Cas9 was proven to be effective in S. boulardii.[9] Boulard
first isolated this yeast after he observed natives of Southeast Asia chewing on the skin of lychee and mangosteen in an attempt to control the symptoms of cholera. In healthy patients, S. boulardii has been shown to be nonpathogenic and nonsystemic (it remains in the gastrointestinal tract rather than spreading elsewhere in the body).
|
| Prokinetic Agent |
Cisapride Monohydrate
https://en.wikipedia.org/wiki/Cisapride

Cisapride is a gastroprokinetic agent, a drug that increases motility in the upper gastrointestinal tract. It acts directly as a serotonin 5-HT4 receptor agonist and
indirectly as a parasympathomimetic. Stimulation of the serotonin receptors increases acetylcholine release in the enteric nervous system. It has been sold under the trade names Prepulsid (Janssen-Ortho) and Propulsid (in the United States). It was discovered by Janssen
Pharmaceutica in 1980. In many countries, it has been either withdrawn from the market or had its indications limited due to incidences of serious cardiac side-effects.
The commercial preparations of this drug are the racemic mixture of both enantiomers of the compound. The (+) enantiomer itself has the major pharmacologic effects and does not induce many of the detrimental side-effects of the mixture.
|
Mosapride citrate
https://en.wikipedia.org/wiki/Mosapride
Mosapride is
a gastroprokinetic agent that
acts as a selective 5HT4 agonist.
The major active metabolite
of mosapride, known as M1, additionally acts as a 5HT3 antagonist, which
accelerates gastric emptying throughout
the whole of the gastrointestinal tract in humans, and
is used for the treatment of gastritis, gastroesophageal
reflux disease, functional
dyspepsia and irritable
bowel syndrome. It
is recommended to be taken on an empty stomach (i.e. at least one hour before food or two hours after food). |
| |
|
| Antibiotics |
| Anthelmintics |
Albendazole
https://en.wikipedia.org/wiki/Albendazole

Albendazole,
also known as albendazolum, is
a medication used for the treatment of a variety of parasitic worm infestations. It
is useful for giardiasis, trichuriasis, filariasis, neurocysticercosis, hydatid
disease, pinworm
disease, and ascariasis,
among other diseases It
is taken by mouth.
As a vermicide, albendazole causes degenerative alterations in the intestinal cells of the worm by binding to the colchicine-sensitive site of β-tubulin, thus inhibiting its polymerization or assembly into microtubules (it binds much better to the β-tubulin of parasites
than that of mammals). Albendazole leads to impaired uptake of glucose by the larval and adult stages of the susceptible parasites, and depletes their glycogen stores. Albendazole also prevents the formation of spindle fibers needed for cell division, which in turn blocks egg production and development; existing eggs are prevented from hatching.Cell motility, maintenance of cell shape, and intracellular transport are also disrupted. At higher concentrations, it disrupts the helminths' metabolic pathways by inhibiting metabolic enzymes such as malate dehydrogenase and fumarate
reductase, with inhibition of the latter leading to less energy produced by the Krebs cycle.Due to diminished ATP production, the parasite is immobilized and eventually dies.
Some parasites have evolved to have some resistance to albendazole by having a different set of acids comprising β-tubulin, decreasing the binding affinity of albendazole.Drosophilia have many of the same mutations, meaning the drug does not affect fruit flies.
|
Febantel
https://en.wikipedia.org/wiki/Fenbendazole

Fenbendazole is
a broad spectrum benzimidazole anthelmintic used
against gastrointestinal parasites including: giardia, roundworms, hookworms, whipworms,
the tapeworm genus Taenia (but
not effective against Dipylidium caninum,
a common dog tapeworm), pinworms, aelurostrongylus, paragonimiasis, strongyles,
and strongyloides that
can be administered to sheep, cattle, horses, fish, dogs, cats, rabbits,
and seals.Fenbendazole is metabolized in
the liver to oxfendazole, which is anthelmintic too; oxfendazole partially
gets reduced back to fenbendazole in the liver and rumen.Also,
fenbendazole itself is an active metabolite of another anthelmintic drug, febantel.
|
Mebendazole
https://en.wikipedia.org/wiki/Mebendazole

Mebendazole (MBZ) is a medication used to treat a number of parasitic worm infestations.This includes ascariasis, pinworm
disease, hookworm infections, guinea worm infections, hydatid disease, and giardia, among others.It is taken by mouth.
Mebendazole is usually well tolerated.Common side effects include headache, vomiting, and ringing in the ears0">ringing .If used at large doses it may cause bone
marrow suppressionone marrow suppression.[3]< It is unclear if it is safe in pregnancy. Mebendazole is a broad-spectrum antihelminthic agent of the benzimidazole type.
Mebendazole works by selectively inhibiting the synthesis ofiting the synthesis of microtubules via binding to colchicine binding site of β-tubulin, thereby blocking polymerisation of tubulin Disruption of cytoplasmic microtubules leads to blocking the
uptake of glucose and other nutrients, resulting in the gradual immobilization and eventual death of the helminths. Poor absorption in digestive tract makes mebendazole an efficient drug for treating intestinal parasitic infections with limited adverse effects. However mebendazole has impact on mammalian cells mostly by inhibiting polymeration ofbiting polymeration of tubulin dimers, thereby disrupting essential microtubule structures such as mito. Disassembly of mitotic spindle then leads to apoptosis mediated via dephosphorylation of Bcl-2 which allows pro-apoptotic protein Bax to dimerize and innitiate programmed cell death.
|
Nitazoxanide
https://en.wikipedia.org/wiki/Nitazoxanide

Nitazoxanide is a broad-spectrum antiparasitic and broad-spectrum antiviral drug that is used in medicine for the treatment of various helminthic, protozoal,
and viral infections. It is indicated for the treatment of infection by Cryptosporidium parvum and Giardia lamblia in immunocompetent individuals and has been repurposed for
the treatment of influenza. Nitazoxanide has also been shown to have in vitro antiparasitic activity and clinical treatment efficacy for infections caused by other protozoa and helminths; emerging evidence suggests that it possesses efficacy in treating a number of viral infections as well.
Chemically, nitazoxanide is the prototype member of the thiazolides, a class of drugs which are synthetic nitrothiazolyl-salicylamide derivatives with antiparasitic and antiviral activity.Tizoxanide,
an active metabolite of nitazoxanide in humans, is also an antiparasitic drug of the thiazolide class.
The anti-protozoal activity of nitazoxanide is believed to be due to interference with the pyruvate:ferredoxin oxidoreductase (PFOR) enzyme-dependent electron transfer reaction which is essential to anaerobic energy metabolism. PFOR inhibition may also contribute to its activity against anaerobic bacteria.
It has also been shown to have activity against influenza A virus in vitro. The mechanism appears to be by selectively blocking the maturation of the viral hemagglutinin at a stage preceding resistance to endoglycosidase H digestion. This impairs hemagglutinin intracellular trafficking and insertion of the protein into the host plasma membrane.
Nitazoxanide modulates a variety of other pathways in vitro, including glutathione-S-transferase and glutamate-gated chloride ion channels in nematodes, respiration and other pathways in bacteria and cancer cells, and viral and host transcriptional factors.
|
| Antifungals |
Anidulafungin
ttps://en.wikipedia.org/wiki/Anidulafungin

Anidulafungin (INN) (trade
names Eraxis, Ecalta)
is a semisynthetic echinocandin used
as an antifungal drug.
It was previously known as LY303366.[3][4][5] It
may also have application in treating invasive Aspergillus infection
when used in combination with Voriconazole.It is a member of the class of antifungal drugs known as the echinocandins;
its mechanism of action is by inhibition of (1→3)-β-D-glucan synthase,
an enzyme important to the synthesis of the fungal cell wall.Anidulafungin inhibits glucan
synthase, an enzyme important in the formation of (1→3)-β-D-glucan,
a major fungal cell wall component. Glucan synthase is not present in mammalian cells, so it is an attractive target for antifungal activity.
|
Eberconazole
https://en.wikipedia.org/wiki/Eberconazole

Eberconazole is
an antifungal drug.
As a 1% topical cream,
it is an effective treatment for dermatophytosis, candidiasis,
and pityriasis.It was approved
for use in Spain in 2015 and is sold under the trade name Ebernet.
|
Fluconazole
https://en.wikipedia.org/wiki/Fluconazole

Fluconazole is
an antifungal medication used
for a number of fungal infections.This
includes candidiasis, blastomycosis, coccidiodomycosis, cryptococcosis, histoplasmosis, dermatophytosis,
and pityriasis versicolor.It
is also used to prevent candidiasis in those who are at high risk such as following organ transplantation,
low birth weight babies, and those with low blood neutrophil counts. It
is given either by mouth or by injection into a vein.
Like
other imidazole-
and triazole-class
antifungals, fluconazole inhibits the fungal cytochrome P450 enzyme 14α-demethylase.
Mammalian demethylase activity is much less sensitive to fluconazole than fungal demethylase. This inhibition prevents the conversion of lanosterol to ergosterol,
an essential component of the fungal cytoplasmic membrane,
and subsequent accumulation of 14α-methyl sterols.Fluconazole is primarily fungistatic; however, it may be fungicidal against
certain organisms in a dose-dependent manner, specifically Cryptococcus.
|
Ketoconazole
https://en.wikipedia.org/wiki/Ketoconazole

Ketoconazole,
sold under the brand name Nizoral among
others, is an antifungal medication
used to treat a number of fungal infections. Applied
to the skin it is used for fungal skin infections such
as tinea, cutaneous
candidiasis, pityriasis
versicolor, dandruff,
and seborrheic dermatitis.Taken by
mouth it is a less
preferred option and only recommended for severe infections when other agents cannot be used. Other
uses include in the treatment of excessive hair growth and Cushing's
syndrome.As
an antifungal, ketoconazole is structurally similar to imidazole,
and interferes with the fungal synthesis of ergosterol,
a constituent of fungal cell membranes,
as well as certain enzymes.
As with all azole antifungal agents, ketoconazole works principally by inhibiting the enzyme cytochrome
P450 14α-demethylase (CYP51A1). This
enzyme participates in the sterol biosynthesis pathway
that leads from lanosterol to ergosterol. |
Luliconazole
https://en.wikipedia.org/wiki/Luliconazole

Luliconazole,
trade names Luzu among
others, is an imidazole antifungal medication.
As a 1% topical cream,
It is indicated for the treatment of athlete's foot, jock
itch, and ringworm caused
y dermatophytes such
as Trichophyton rubrum, Microsporum
gypseum and Epidermophyton
floccosum.
|
Micafungin
https://en.wikipedia.org/wiki/Micafungin

Micafungin (trade
name Mycamine) is an echinocandin antifungal
drug used to treat and
prevent invasive fungal infections including candidemia, abscesses and esophageal candidiasis. It inhibits the production of beta-1,3-glucan,
an essential component of fungal cell walls.
Micafungin is administered intravenously.
It received final approval from the U.S. Food and Drug Administration on
March 16, 2005, and gained approval in the European Union on
April 25, 2008.
|
Sodium Posaconazole
https://en.wikipedia.org/wiki/Posaconazole
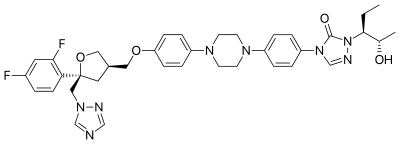
Posaconazole, sold under the brand names Noxafil and Posanol is a triazole antifungal medication.
It was approved for medical use in the United States in September 2006, and is available as a generic medication.
Posaconazole works by disrupting the close packing of acyl chains of phospholipids,
impairing the functions of certain membrane-bound enzyme systems such as ATPase and enzymes of the electron transport system, thus inhibiting growth of the fungi. It does this by blocking the synthesis of ergosterol by
inhibiting of the enzyme lanosterol 14α-demethylase and
accumulation of methylated sterol precursors. Posaconazole is significantly more potent at inhibiting 14-alpha demethylase than itraconazole.
|
Tavaborole
https://en.wikipedia.org/wiki/Tavaborole

Tavaborole,
sold under the brand name Kerydin, is a topical antifungal
medication for the
treatment of onychomycosis,
a fungal infection of
the nail and nail
bed. Tavaborole began phase
III clinical trials in
December 2010 and was
approved by the US FDA in July 2014.The medication inhibits an essential fungal enzyme, leucyl-tRNA
synthetase, that is required for protein
synthesis. The inhibition of protein synthesis leads to termination of cell
growth and then cell
death, eliminating the fungal infection.
Tavaborole, when prepared with a 1:1 mixture of ethyl acetate and propylene glycol, has the ability to fully penetrate through the human nail.[citation needed] In studies with cadaver
fingernails, a 5% solution of tavaborole penetrated the nail an average of 524.7 mcg/cm2 after two weeks of daily use.
Tavaborole is detectable in the blood at a level of 3.54 ng/mL after a single use of 0.2 mL of the 5% solution. Tavaborole has an elimination half-life of 28.5 hours, a maximum concentration of 5.17 ng/mL after two weeks of daily use, and takes 8 days to reach the maximum concentration.
|
Itraconazole
https://en.wikipedia.org/wiki/Itraconazole

Itraconazole is
an antifungal medication used
to treat a number of fungal infections. This
includes aspergillosis, blastomycosis, coccidioidomycosis, histoplasmosis,
and paracoccidioidomycosis.
The mechanism of action of itraconazole is the same as the other azole antifungals: it inhibits the fungal-mediated synthesis of ergosterol, via inhibition of lanosterol 14α-demethylase. Because of its ability to inhibit cytochrome P450 3A4 CC-3, caution should be used when considering
interactions with other medications.
Itraconazole is pharmacologically distinct from other azole antifungal agents in that it is the only inhibitor in this class that has been shown to inhibit both the hedgehog signaling pathway and angiogenesis These distinct activities are unrelated to inhibition of the cytochrome
P450 lanosterol 14 alpha-demethylase and the exact molecular targets responsible remain unidentified. Functionally, the antiangiogenic activity of itraconazole has been shown to be linked to inhibition of glycosylation, VEGFR2 phosphorylation, trafficking, and cholesterol biosynthesis pathways. Evidence suggests the structural determinants for inhibition of hedgehog signaling by itraconazole are recognizably different from those associated with antiangiogenic activity.
|
| Antimalarial Drug |
Lumefantrine
https://en.wikipedia.org/wiki/Lumefantrine

Lumefantrine (or benflumetol)
is an antimalarial drug.
It is only used in combination with artemether.
The term "co-artemether" is sometimes used to describe this combination. Lumefantrine
has a much longer half-life compared to artemether, and is therefore thought to clear any residual parasites that remain after combination treatment.
|
| Antiretroviral |
Abacavir Sulphate
https://en.wikipedia.org/wiki/Abacavir

Abacavir,
sold under the brand name Ziagen, is a medication used
to prevent and treat HIV/AIDS.[1][2] Similar
to other nucleoside analog reverse-transcriptase
inhibitors (NRTIs),
abacavir is used together with other HIV medications,
and is not recommended by itself. It
is taken by mouth as a tablet or solution and may be used in children over the age of three months.Abacavir is a nucleoside
reverse transcriptase inhibitor that
inhibits viral replication. It is a guanosine analogue
that is phosphorylated to carbovir triphosphate (CBV-TP). CBV-TP competes with the viral molecules and is incorporated into the viral DNA. Once CBV-TP is integrated into the viral DNA, transcription and
HIV reverse transcriptase is inhibited.
|
Atazanavir
https://en.wikipedia.org/wiki/Atazanavir

Atazanavir,
sold under the trade name Reyataz among
others, is an antiretroviral medication used
to treat and prevent HIV/AIDS. It
is generally recommended for use with other antiretrovirals.It may be used for prevention after a needlestick injury or
other potential exposure. It
is taken by mouth once a day..Atazanavir binds to the active site HIV protease and prevents it from cleaving the pro-form of viral proteins into the working machinery of the virus.If the HIV protease enzyme does not work, the virus is not infectious, and no mature virions are made.The azapeptide drug was designed as
an analog of the peptide chain substrate that HIV protease would cleave normally into active viral proteins. More specifically, atazanavir is a structural analog of the transition state during which the bond between a phenylalanine and proline is broken.Humans do not have any enzymes that break bonds between phenylalanine and proline, so this drug will not target human enzymes.
|
Efavirenz
https://en.wikipedia.org/wiki/Efavirenz

Efavirenz (EFV),
sold under the brand names Sustiva among
others, is an antiretroviral medication used
to treat and prevent HIV/AIDS.It
is generally recommended for use with other antiretrovirals.It may be used for prevention after a needlestick
injury or other potential
exposure.It is sold both by itself and in combination as efavirenz/emtricitabine/tenofovir..
Efavirenz falls in the NNRTI class of antiretrovirals. Both nucleoside and non-nucleoside RTIs inhibit the same target, the reverse transcriptase enzyme, an essential viral enzyme which transcribes viral RNA into DNA. Unlike nucleoside RTIs, which bind at the enzyme's active site, NNRTIs act allosterically by binding to a distinct site away from the active site known as the NNRTI pocket.
Efavirenz is not effective against HIV-2, as the pocket of the HIV-2 reverse transcriptase has a different structure, which confers intrinsic resistance to the NNRTI class.
As most NNRTIs bind within the same pocket, viral strains which are resistant to efavirenz are usually also resistant to the other NNRTIs, nevirapine and delavirdine. The most common mutation observed after efavirenz treatment is K103N, which is also observed with other NNRTIs. Nucleoside reverse-transcriptase inhibitors (NRTIs) and efavirenz have different binding targets, so cross-resistance
is unlikely; the same is true with regard to efavirenz and protease inhibitors.
|
Darunavir (Amorphous / Ethanolate / Sodium)
https://en.wikipedia.org/wiki/Darunavir

Darunavir (DRV),
sold under the brand name Prezista among
others, is an antiretroviral medication used
to treat and prevent HIV/AIDS.[1] It
is generally recommended for use with other antiretrovirals.It is often used with low doses of ritonavir or cobicistat to
increase darunavir levels.Darunavir is a nonpeptidic inhibitor of protease (PR) that lodges itself in the active site of PR
through a number of hydrogen bonds.It was developed to increase interactions with HIV-1 protease and
to be more resistant against HIV-1 protease mutations. With a Kd (dissociation
constant) of 4.5 x
10−12 M,
darunavir has a much stronger interaction with PR and its dissociation constant is 1/100 to 1/1000 of other protease inhibitors.This strong interaction comes from increased hydrogen bonds between darunavir and the backbone of the PR active site (Figure 2). Darunavir's structure allows it to create more hydrogen bonds with the PR active site than most PIs that have been developed and approved by the FDA Furthermore, the backbone of HIV-1 protease maintains its spatial conformation in the presence of mutations.Because darunavir interacts with this stable portion of the protease, the PR-PI interaction is less likely to be disrupted by a mutation.
|
Indinavir Sulphate
https://en.wikipedia.org/wiki/Indinavir

Indinavir (IDV;
trade name Crixivan, made by Merck)
is a protease inhibitor used
as a component of highly active antiretroviral therapy to
treat HIV/AIDS. It is soluble white
powder administered orally in combination with other antiviral drugs. The drug prevents protease from functioning normally. Consequently, HIV viruses cannot reproduce, causing a decrease in the viral load. Commercially sold indinavir is indinavir anhydrous, which is indinavir with an additional amine in the hydroxyethylene backbone. This enhances its solubility and oral bioavailability, making it easier for users to intake. It was synthetically produced for the purpose of inhibiting the protease in the HIV virus.
|
Oseltamivir
https://en.wikipedia.org/wiki/Oseltamivir

Oseltamivir,
sold under the brand name Tamiflu, is an antiviral
medication used to treat
and prevent influenza A and influenza
B (flu).Many
medical organizations recommend it in people who have complications or are at high risk of complications within 48 hours of first symptoms of infection.They recommend it to prevent infection in those at high risk, but not the general population.Oseltamivir is a neuraminidase
inhibitor, a competitive inhibitor of influenza's neuraminidase enzyme.
The enzyme cleaves the sialic acid which is found on glycoproteins on
the surface of human cells that helps new virions to exit the cell. Thus oseltamivir prevents new viral particles from being released.
|
Rilpivirine HCl
https://en.wikipedia.org/wiki/Rilpivirine

Rilpivirine (TMC278,
trade name Edurant) is a pharmaceutical
drug, developed by Tibotec,
for the treatment of HIV infection.It
is a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI)
with higher potency,
longer half-life and
reduced side-effect profile compared
with older NNRTIs, such as efavirenz.
|
Ritonavir
https://en.wikipedia.org/wiki/Ritonavir

Ritonavir (RTV),
sold under the brand name Norvir, is an antiretroviral
medication used along with
other medications to treat HIV/AIDS.[1] This
combination treatment is known as highly active antiretroviral therapy (HAART).Often
a low dose is used with other protease inhibitors.It
may also be used in combination with other medications for hepatitis C.It
is taken by mouth.The
capsules of the medication do not work the same as the tablets.
Ritonavir was originally developed as an inhibitor of HIV protease, one of a family of pseudo-C2-symmetric small molecule inhibitors.[citation
needed]
Ritonavir is now rarely used for its own antiviral activity but remains widely used as a booster of other protease inhibitors. More specifically, ritonavir is used to inhibit a particular enzyme, in intestines, liver, and elsewhere, that normally metabolizes protease inhibitors, cytochrome P450-3A4 (CYP3A4).The drug binds to and inhibits CYP3A4, so a low dose can be used to
enhance other protease inhibitors. This discovery drastically reduced the adverse effects and improved the efficacy of protease inhibitors and HAART. However, because of the general role of CYP3A4 in xenobiotic metabolism, dosing with ritonavir also affects the efficacy of numerous other medications, adding to the challenge of prescribing drugs concurrently.
|
Saquinavir
https://en.wikipedia.org/wiki/Saquinavir

Saquinavir (SQV),
sold under the brand names Invirase and Fortovase,
is an antiretroviral drug used
together with other medications to treat or prevent HIV/AIDS.Typically
it is used with ritonavir or lopinavir/ritonavir to
increase its effect.It is taken by mouth.Saquinavir binds to the active site of the viral protease and prevents cleavage of viral polyproteins, preventing maturation of the virus. Saquinavir inhibits both HIV-1 and HIV-2 proteases.
|
Simeprevir
https://en.wikipedia.org/wiki/Simeprevir

Simeprevir,
sold under the trade names Olysio among
others, is a medication used in combination with other medications for the treatment of hepatitis C.It
is specifically used for hepatitis C genotype 1 and 4.Medications it is used with include sofosbuvir or ribavirin and peginterferon-alfa.Cure
rates are in 80s to 90s percent.It may be used in those who also have HIV/AIDS.It
is taken by mouth once daily for typically 12 weeks..Simeprevir is a NS3/4A protease inhibitor, thus preventing viral maturation through inhibition of protein synthesis. Simeprevir is administered as one capsule once daily with pegylated interferon and ribavirin for the treatment of genotype 1 or genotype 4 chronic hepatitis C in adult people with compensated liver disease (including cirrhosis), with or without HIV-1 co-infection, who are treatment naive or who have failed previous interferon
therapy.Genotype 1 is the most prevalent form of hepatitis C virus (HCV) worldwide.
|
Tenofovir
https://en.wikipedia.org/wiki/Tenofovir_disoproxil

Tenofovir disoproxil,
sold under the trade name Viread among
others, is a medication used to treat chronic hepatitis B and
to prevent and treat HIV/AIDS.It
is generally recommended for use with other antiretrovirals.It may be used for prevention of HIV/AIDS among those at high risk before exposure, and after a needlestick
injury or
other potential exposure.It is sold both by itself and together as emtricitabine/tenofovir and efavirenz/emtricitabine/tenofovir.It
does not cure HIV/AIDS or hepatitis B.It is available by mouth as a tablet or powder.enofovir
disoproxil is a nucleotide
analog reverse-transcriptase inhibitor (NtRTI).It
selectively inhibits viral reverse transcriptase,
a crucial enzyme in retroviruses such as human immunodeficiency virus (HIV),
while showing limited inhibition of human enzymes, such as DNA polymerases α,
β, and mitochondrial DNA polymerase
γ.In vivo tenofovir disoproxil fumarate is converted to tenofovir, an acyclic analog of deoxyadenosine 5'-monophosphate (d-AMP). Tenofovir lacks a hydroxyl group in the position corresponding to the 3' carbon of the d-AMP, preventing the formation of the 5′ to 3′ phosphodiester linkage
essential for DNA chain elongation.Once incorporated into a growing DNA strand, tenofovir causes premature termination of DNA transcription, preventing viral.
|
| Antivirals |
Acyclovir

Aciclovir (ACV),
also known as acyclovir, is an antiviral
medication. It is primarily used for the treatment of herpes
simplex virus infections, chickenpox,
and shingles. Other uses include
prevention of cytomegalovirus infections
following transplant and severe complications of Epstein-Barr virus infection.It
can be taken by mouth, applied as a cream, or injected.Aciclovir
is converted by viral thymidine kinase to
aciclovir monophosphate, which is then converted by host cell kinases to aciclovir triphosphate (ACV-TP). ACV-TP, in turn, competitively inhibits and
inactivates HSV-specified DNA
polymerases preventing further viral DNA synthesis without affecting the normal cellular processes.
|
Lamivudine
https://en.wikipedia.org/wiki/Lamivudine

Lamivudine,
commonly called 3TC, is an antiretroviral
medication used to prevent
and treat HIV/AIDS. It
is also used to treat chronic hepatitis B when
other options are not possible. It
is effective against both HIV-1 and HIV-2. It
is typically used in combination with other antiretrovirals such as zidovudine and abacavir. Lamivudine
may be included as part of post-exposure prevention in
those who have been potentially exposed to HIV. Lamivudine
is taken by mouth as a liquid or tablet.
Lamivudine is an analogue of cytidine. It can inhibit both types (1 and 2) of HIV reverse transcriptase and also the reverse transcriptase of hepatitis
B virus. It is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The lack of a 3'-OH group in the incorporated nucleoside analogue prevents the formation of the 5' to 3' phosphodiester linkage essential for DNA chain elongation, and therefore, the viral DNA growth is terminated.
Lamivudine is administered by mouth, and it is rapidly absorbed with a bio-availability of over 80%. Some research suggests that lamivudine can cross the blood–brain barrier. Lamivudine is often given in combination with zidovudine, with which it is highly synergistic. Lamivudine treatment has been shown to restore zidovudine sensitivity of previously resistant HIV. Lamivudine showed no evidence of carcinogenicity or mutagenicity in in
vivo studies in mice and rats at doses from 10 to 58 times those used in humans.
|
Valacyclovir
https://en.wikipedia.org/wiki/Valaciclovir

Valaciclovir,
also spelled valacyclovir, is an antiviral
medication used to treat
outbreaks of herpes simplex or herpes
zoster (shingles).It
is also used to prevent cytomegalovirus following
a kidney transplant in
high risk cases. It is taken by mouth.
Aciclo-GTP, the active triphosphate metabolite of aciclovir, is a very potent inhibitor of viral DNA replication. Aciclo-GTP competitively inhibits and inactivates the viral DNA polymerase.Its monophosphate form also incorporates into the viral DNA, resulting in chain
termination. It has also been shown that the viral enzymes cannot remove aciclo-GMP from the chain, which results in inhibition of further activity of DNA polymerase. Aciclo-GTP is fairly rapidly metabolized within the cell, possibly by cellular phosphatases.Aciclovir is active against most species in the herpesvirus family.
|
Valganciclovir
https://en.wikipedia.org/wiki/Valganciclovir

Valganciclovir,
sold under the brandname Valcyte among
others, is an antiviral medication used
to treat cytomegalovirus (CMV) infection in
those with HIV/AIDS or
following organ transplant.[1] It
is often used long term as it only suppresses rather than cures the infection. Valganciclovir
is taken by mouth.[
Valganciclovir is a prodrug for ganciclovir, which is a synthetic analog of 2′-deoxy-guanosine. Its structure is the same as ganciclovir, except for the addition of a L-valyl ester at the 5' end of the incomplete deoxyribose ring. The valine increases both the absorption of the drug in the intestines, as well as the bioavailability of the drug once it is absorbed. The L-valyl ester is cleaved by esterases in
the intestines and the liver, leaving ganciclovir to be absorbed by the virus-infected cells.
Ganciclovir is first phosphorylated to ganciclovir monophosphate by a viral thymidine kinase encoded by the cytomegalovirus (CMV) upon infection. Human cellular kinases further phosphorylate the molecule to create ganciclovir diphosphate, then ganciclovir triphosphate. These kinases are present in 10-fold greater concentrations in CMV or herpes simplex virus (HSV)-infected cells compared to uninfected human cells, allowing ganciclovir triphosphate to concentrate in infected cells.
Ganciclovir triphosphate is a competitive inhibitor of deoxyguanosine triphosphate (dGTP). It is incorporated into viral DNA and preferentially inhibits viral DNA polymerases more than cellular DNA polymerases. Ganciclovir triphosphate serves as a poor substrate for chain elongation. Once incorporated into viral DNA due to its structure similarity to dGTP, chain termination occurs once a single nucleotide is added to the distal hydroxyl group of the incomplete deoxyribose ring of ganciclovir.
|
| Beta-Lactam |
Faropenem Sodium
https://en.wikipedia.org/wiki/Faropenem

Faropenem is
an orally active beta-lactam antibiotic belonging
to the penem group. It
is resistant to some forms of extended-spectrum beta-lactamase. It
is available for oral use. |
| Carbapenem |
Doripenem
https://en.wikipedia.org/wiki/Doripenem

Doripenem (Doribax, Finibax) is an antibiotic drug in the carbapenem class. It is a beta-lactam antibiotic drug able to kill Pseudomonas aeruginosa.
Doripenem can be used for bacterial infections such as: complex abdominal infections, pneumonia within the setting of a hospital, and complicated infections of the urinary tract including kidney infections with sepsis.
The greater stability of doripenem in aqueous solution compared to earlier members of the carbapenem class allows it to be administered as an infusion over 4 hours or more, which may be advantageous in the treatment of certain difficult-to-treat infections. It may present a lower risk of inducing seizures than other carbapenems.
|
Ertapenem
https://en.wikipedia.org/wiki/Ertapenem

Ertapenem (trade
name Invanz) is a carbapenem antibiotic medication
for the treatment of infections of the abdomen,
the lungs, the upper part of the female reproductive system,
and diabetic foot, used in
the form of infusions or injections.Like all beta-lactam antibiotics, ertapenem is bactericidal. It
inhibits cross-linking of the peptidoglycan layer of bacterial cell walls by blocking a type of enzymes called penicillin-binding
proteins (PBPs). When a bacterial cell tries to synthesize new cell wall in order to grow and divide, the attempt fails, rendering the cell vulnerable to osmotic disruption. Additionally, the surplus of peptidoglycan precursors triggers autolytic enzymes
of the bacterium, which disintegrate the existing wall.
|
Imipenem with cilastatin (Imipenem/cilastatin)
https://en.wikipedia.org/wiki/Imipenem/cilastatin

Imipenem/cilastatin,
sold under the brand name Primaxin among
others, is an antibiotic useful
for the treatment of a number of bacterial infections. It
is made from a combination of imipenem and cilastatin. Specifically
it is used for pneumonia, sepsis, endocarditis, joint
infections, intra-abdominal
infections, and urinary
tract infections. It
is given by injection into a vein or muscle.
Imipenem/cilastatin has the ability to kill a wide variety of bacteria. Imipenem is the active antibiotic agent and works by interfering with their ability to form cell walls, so the bacteria break up and die.
Imipenem is rapidly degraded by the renal enzyme dehydropeptidase if administered alone (making it less effective); the metabolites can cause kidney damage. Imipenem is a broad-spectrum betalactam antibiotic used for severe bacterial infections caused by susceptible organisms. Because imipenem is rapidly inactivated by renal dehydropeptidase I, it is given in combination with cilastatin, a DHP-I inhibitor which increases half-life and tissue penetration of imipenem.
|
Meropenem Trihydrate
https://en.wikipedia.org/wiki/Meropenem

Meropenem,
sold under the brandname Merrem among
others, is a broad-spectrum antibiotic used
to treat a variety of bacterial infections. Some
of these include meningitis, intra-abdominal
infection, pneumonia, sepsis,
and anthrax. It
is given by injection into a vein.
Meropenem is bactericidal except against Listeria monocytogenes, where it is bacteriostatic. It inhibits bacterial cell wall synthesis like other β-lactam antibiotics. In contrast to other beta-lactams, it is highly resistant to degradation by β-lactamases or
cephalosporinases. In general, resistance arises due to mutations in penicillin-binding proteins, production of metallo-β-lactamases, or resistance to diffusion across the bacterial outer membrane. Unlike imipenem, it is stable to dehydropeptidase-1, so can be given without cilastatin.
In 2016, a synthetic peptide-conjugated PMO (PPMO) was found to inhibit the expression of New Delhi metallo-beta-lactamase, an enzyme that many drug-resistant bacteria use to destroy carbapenems.
|
| Drugs used in Ornidazole Protozoal Infections |
Tinidazole
https://en.wikipedia.org/wiki/Tinidazole

Tinidazole is
a drug used against protozoan infections.
It is widely known throughout Europe and the developing world as a treatment for a variety of amoebic and parasitic infections.
It was developed in 1972 and is a prominent member of the nitroimidazole antibiotic class.A
large body of clinical data exists to support use of tinidazole for infections from amoebae, giardia, and trichomonas,
just like metronidazole. Tinidazole may be a therapeutic alternative in the setting of metronidazole intolerance. Tinidazole may also be used to treat a variety of other bacterial infections
(e.g., as part of combination therapy for Helicobacter
pylori eradication protocols).
|
Metronidazole Benzoate
https://en.wikipedia.org/wiki/Metronidazole

Metronidazole, marketed under the brand name Flagyl among
others, is an antibiotic and antiprotozoal
medication. It
is used either alone or with other antibiotics to treat pelvic inflammatory disease, endocarditis,
and bacterial vaginosis. It
is effective for dracunculiasis, giardiasis, trichomoniasis,
and amebiasis. It
is an option for a first episode of mild-to-moderate Clostridium difficile colitis if vancomycin or fidaxomicin is
unavailable.
Metronidazole is of the nitroimidazole class.
It inhibits nucleic acid synthesis by forming nitroso radicals,
which disrupt the DNA of microbial cells. This
function only occurs when metronidazole is partially reduced, and because this reduction usually happens only in anaerobic bacteria and protozoans, it has relatively little effect upon human cells or aerobic bacteria. |
| Glycopeptide Antibacterial |
Teicoplanin
https://en.wikipedia.org/wiki/Teicoplanin

Teicoplanin is
an antibiotic used
in the prophylaxis and
treatment of serious infections caused by Gram-positive bacteria,
including methicillin-resistant Staphylococcus aureus and Enterococcus
faecalis. It is a semisynthetic glycopeptide
antibiotic with a spectrum
of activity similar to vancomycin.
Its mechanism of action is to inhibit bacterial cell wall synthesis.Teicoplanin (TARGOCID, marketed by Sanofi Aventis Ltd) is actually a mixture of several compounds, five major (named teicoplanin
A2-1 through A2-5)
and four minor (named teicoplanin RS-1 through RS-4). All
teicoplanins share a same glycopeptide core,
termed teicoplanin A3-1 — a fused
ring structure to which two carbohydrates (mannose and N-acetylglucosamine)
are attached. The major and minor components also contain a third carbohydrate moiety — β-D-glucosamine —
and differ only by the length and conformation of a side-chain attached to it.
|
| Macrolides |
Azithromycin
https://en.wikipedia.org/wiki/Azithromycin

Azithromycin is
an antibiotic used
for the treatment of a number of bacterial infections. This
includes middle ear infections, strep
throat, pneumonia, traveler's
diarrhea, and certain other intestinal
infections. It
can also be used for a number of sexually transmitted infections,
including chlamydia and gonorrhea
infections. Along
with other medications, it may also be used for malaria. It
can be taken by mouth or intravenously with
doses once per day.Azithromycin prevents bacteria from growing by interfering with their protein
synthesis. It binds to the 50S subunit of the bacterial ribosome, thus inhibiting translation of mRNA.
Nucleic acid synthesis is not affected.
|
Erythromycin
https://en.wikipedia.org/wiki/Erythromycin
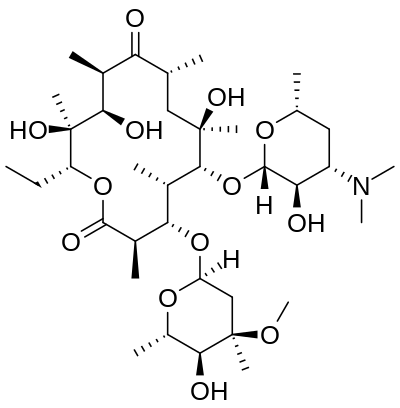
Erythromycin is
an antibiotic used
for the treatment of a number of bacterial infections. This
includes respiratory tract infections, skin
infections, chlamydia
infections, pelvic
inflammatory disease, and syphilis. It
may also be used during pregnancy to
prevent Group B streptococcal infection in
the newborn, as well as
to improve delayed stomach emptying. It
can be given intravenously and
by mouth. An eye
ointment is routinely recommended after delivery to prevent eye infections in the newborn.
Erythromycin displays bacteriostatic activity or inhibits growth of bacteria, especially at higher concentrations. By binding to the 50s subunit of the bacterial rRNA complex, protein synthesis and subsequent structure and function processes critical for life or replication are inhibited. Erythromycin interferes with aminoacyl translocation, preventing the transfer of the tRNA bound
at the A site of the rRNA complex to the P site of the rRNA complex. Without this translocation, the A site remains occupied, thus the addition of an incoming tRNA and its attached amino acid to the nascent polypeptide chain is inhibited. This interferes with
the production of functionally useful proteins, which is the basis of this antimicrobial action.
Erythromycin increases gut motility by binding to Motillin, thus it is a Motillin receptor agonist in addition to its antimicrobial properties.
|
Fidaxomicin
https://en.wikipedia.org/wiki/Fidaxomicin

Fidaxomicin,
sold under the brand name Dificid among
others, is the first member of a class of narrow spectrum macrocyclic antibiotic drugs
called tiacumicins.[2] It
is a fermentation product obtained from the actinomycete Dactylosporangium aurantiacum subspecies hamdenesis. Fidaxomicin
is minimally absorbed into the bloodstream when taken orally, is bactericidal,
and selectively eradicates pathogenic Clostridium
difficile with
relatively little disruption to the multiple species of bacteria that
make up the normal, healthy intestinal flora.
The maintenance of normal physiological conditions in the colon may reduce the probability of recurrence of Clostridium difficile infection.Fidaxomicin
binds to and prevents movement of the "switch regions" of bacterial RNA polymerase. Switch motion occurs during the opening and closing of the DNA:RNA clamp, a process that occurs throughout RNA transcription but is especially important in the opening of double-stranded DNA during the initiation of transcription. It
has minimal systemic absorption and a narrow spectrum of activity; it is active against Gram positive bacteria, especially clostridia.
The minimal inhibitory concentration (MIC) range for C. difficile (ATCC 700057) is 0.03–0.25 μg/mL
|
| Oxazolidinones |
Linezolid
https://en.wikipedia.org/wiki/Linezolid
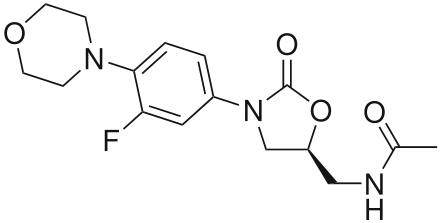
Linezolid is an antibiotic used for the treatment of infections caused by Gram-positive bacteria that are resistant to other antibiotics. Linezolid is active against most
Gram-positive bacteria that cause disease, including streptococci, vancomycin-resistant enterococci (VRE), and methicillin-resistant Staphylococcus aureus (MRSA). The main uses are infections of the skin and pneumonia although
it may be used for a variety of other infections including drug-resistant tuberculosis.It is used either by injection into a vein or by mouth.
When given for short periods, linezolid is a relatively safe antibiotic. It can be used in people of all ages and in people with liver disease or poor kidney function.
Linezolid, like other oxazolidinones, is a bacterial protein
synthesis inhibitor and a
weak, non-selective, reversible monoamine
oxidase inhibitor. As
a protein synthesis inhibitor, linezolid stops the growth and reproduction of bacteria by disrupting translation of messenger
RNA (mRNA) into proteins in
bacterial ribosomes. Linezolid
inhibits translation at the first step of protein synthesis, initiation unlike
most other protein synthesis inhibitors, which inhibit elongation. It
does so by preventing the formation of the initiation complex, composed of the 30S and 50S subunits
of the ribosome, tRNA,
and mRNA. Linezolid binds to the 23S portion
of the 50S subunit (the center of peptidyl transferase activity), close
to the binding sites of chloramphenicol, lincomycin,
and other antibiotics. Due to this unique mechanism of action, cross-resistance between
linezolid and other protein synthesis inhibitors is highly infrequent or nonexistent.
|
| Penicillins |
Amoxycillin
https://en.wikipedia.org/wiki/Amoxicillin
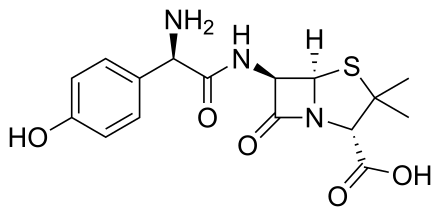
Amoxicillin is
an antibiotic used
to treat a number of bacterial infections. These
include middle ear infection, strep
throat, pneumonia, skin
infections, and urinary
tract infections among
others. It is taken by
mouth, or less commonly by injection.As a derivative of ampicillin,
amoxicillin is a member of the penicillin family and, like penicillins, is a β-lactam
antibiotic. It inhibits cross-linkage between the linear peptidoglycan polymer chains that make up a major component of the bacterial cell
wall. It has two ionizable groups in the physiological range (the amino group in alpha-position to the amide carbonyl group and the carboxyl group). |
Ampicillin
https://en.wikipedia.org/wiki/Ampicillin

Ampicillin is
an antibiotic used
to prevent and treat a number of bacterial infections,
such as respiratory tract infections, urinary
tract infections, meningitis, salmonellosis,
and endocarditis. It
may also be used to prevent group B streptococcal infection in
newborns. It is used by
mouth, by injection into a muscle,
or intravenously.Like all antibiotics, it is not useful for the treatment of viral infections.
Ampicillin is in the penicillin group of beta-lactam antibiotics and is part of the aminopenicillin family. It is roughly equivalent to amoxicillin in terms of activity.[3] Ampicillin
is able to penetrate Gram-positive and some Gram-negative bacteria. It differs from penicillin G, or benzylpenicillin, only by the presence of an amino group. This amino group, present on both ampicillin and amoxicillin, helps these antibiotics pass through the pores of the outer membrane of Gram-negative bacteria, such as E. coli, Proteus mirabilis, Salmonella
enterica, and Shigella.
Ampicillin acts as an irreversible inhibitor of the enzyme transpeptidase, which is needed by bacteria to make the cell wall. It inhibits the third and final stage of bacterial cell wall synthesis in binary fission, which ultimately leads to cell lysis; therefore, ampicillin is usually bacteriolytic.
|
Ampicillin with Sulbactam Sodium
https://en.wikipedia.org/wiki/Sulbactam

Sulbactam is
a β-lactamase inhibitor.
This drug is
given in combination with β-lactam antibiotics to
inhibit β-lactamase,
an enzyme produced
by bacteria that
destroys the antibiotics.Sulbactam
is an irreversible inhibitor of β-lactamase; it binds to the enzyme and does not allow it to degrade the antibiotic
|
Cloxacillin
https://en.wikipedia.org/wiki/Cloxacillin
Cloxacillin is
an antibiotic useful
for the treatment of a number of bacterial infections. This
includes impetigo, cellulitis, pneumonia, septic
arthritis, and otitis
externa. It
is not effective for methicillin-resistant Staphylococcus aureus (MRSA). It
is used by mouth and by injection.It is It is semisynthetic and in the same class as penicillin.
Cloxacillin is used against staphylococci that produce beta-lactamase,
due to its large R chain, which does not allow the beta-lactamases to bind. This drug has a weaker antibacterial activity than benzylpenicillin,
and is devoid of serious toxicity except for allergic reactions.
|
Pienicillin-G-Amidase Enzyme
https://en.wikipedia.org/wiki/Amidase

In enzymology,
an amidase (EC 3.5.1.4, acylamidase, acylase
(misleading), amidohydrolase
(ambiguous), deaminase
(ambiguous), fatty
acylamidase, N-acetylaminohydrolase
(ambiguous)) is an enzyme that catalyzes the hydrolysis of
an amide.
|
Piperacillin with Tazobactam
https://en.wikipedia.org/wiki/Piperacillin

Piperacillin is a broad-spectrum β-lactam antibiotic of the ureidopenicillin class. The chemical structure of piperacillin and other ureidopenicillins incorporates a polar side chain that enhances penetration into gram-negative bacteria and reduces susceptibility to cleavage by gram-negative beta lactamase enzymes. These properties confer activity against
the important hospital pathogen Pseudomonas aeruginosa. Thus piperacillin is sometimes referred to as an "anti-pseudomonal penicillin".
When used alone, piperacillin lacks strong activity against the gram-positive pathogens such as Staphylococcus aureus, as the beta-lactam ring is hydrolyzed by the bacteria's beta-lactamase.
https://en.wikipedia.org/wiki/Tazobactam

Tazobactam is a pharmaceutical drug that inhibits the action of bacterial β-lactamases, especially those belonging to the SHV-1 and TEM groups. It is commonly used as its sodium salt, tazobactam sodium. In simple terms, it is an
ingredient that can be added to certain antibiotics to make them less vulnerable to bacteria's antimicrobial resistance.
Tazobactam is combined with the extended spectrum β-lactam antibiotic piperacillin in the drug piperacillin/tazobactam, used in infections due to Pseudomonas
aeruginosa. Tazobactam broadens the spectrum of piperacillin by making it effective against organisms that express β-lactamase and would normally degrade piperacillin.
Tazobactam is a heavily modified penicillin and a sulfone
|
| Quinolones |
Ciprofloxacin
https://en.wikipedia.org/wiki/Ciprofloxacin

Ciprofloxacin is
an antibiotic used
to treat a number of bacterial infections.This
includes bone and joint infections,
intra abdominal infections, certain type of infectious diarrhea, respiratory
tract infections, skin infections, typhoid
fever, and urinary
tract infections, among others. For
some infections it is used in addition to other antibiotics. It
can be taken by mouth, as eye drops, as ear drops, or intravenously.Ciprofloxacin is a broad-spectrum antibiotic of the fluoroquinolone class.
It is active against some Gram-positive and many Gram-negative bacteria. It
functions by inhibiting a type
II topoisomerase (DNA
gyrase) and topoisomerase IV, necessary to separate bacterial DNA, thereby inhibiting cell division.
|
Garenoxacin
https://en.wikipedia.org/wiki/Garenoxacin

Garenoxacin (INN)
is a quinolone antibiotic for
the treatment of Gram-positive and Gram-negative bacterial
infections.Garenoxacin was discovered by Toyama Chemical Co., Ltd. of Tokyo, Japan, and is currently being marketed in Japan under the tradename Geninax.
|
Levofloxacin
https://en.wikipedia.org/wiki/Levofloxacin
Levofloxacin,
sold under the trade names Levaquin among
others, is an antibiotic. It
is used to treat a number of bacterial infections including acute
bacterial sinusitis, pneumonia, H.
pylori (in combination with
other medications), urinary tract infections, chronic
prostatitis, and some types of gastroenteritis.Along
with other antibiotics it may be used to treat tuberculosis, meningitis,
or pelvic inflammatory disease.Use
is generally recommended only when other options are not available.It is available by mouth, intravenously,and
in eye drop form.
Levofloxacin is a broad-spectrum antibiotic that is active against both Gram-positive and Gram-negative bacteria. Like all quinolones, it functions by inhibiting the DNA gyrase and topoisomerase
IV, two bacterial type II topoisomerases. Topoisomerase IV is necessary to separate DNA that has been replicated (doubled) prior to bacterial cell division. With the DNA not being separated, the process is stopped, and the bacterium cannot divide. DNA gyrase, on the other hand, is responsible for supercoiling the
DNA, so that it will fit in the newly formed cells. Both mechanisms amount to killing the bacterium. Levofloxacin acts as a bactericide.
As of 2011 the mechanism of action for the drug's musculoskeletal complications were not clear.
|
Marbofloxacin
https://en.wikipedia.org/wiki/Marbofloxacin

Marbofloxacin is
a carboxylic acid derivative third generation fluoroquinolone antibiotic.
It is used in veterinary medicine under
the trade names Marbocyl, Forcyl,
Marbo vet and Zeniquin.
A formulation of marbofloxacin combined with clotrimazole and dexamethasone is
available under the name Aurizon (CAS number 115550-35-1).Its
mechanism of action is not thoroughly understood, but it is believed to be similar to the other fluoroquinolones by impairing the bacterial DNA
gyrase which results in rapid bactericidal activity.[1] The
other proposed mechanisms include that it acts against nondividing bacteria and does not require protein and RNA
synthesis, which block protein and RNA
synthesis respectively.
|
Ofloxacin
https://en.wikipedia.org/wiki/Ofloxacin

Ofloxacin is
an antibiotic useful
for the treatment of a number of bacterial infections. When
taken by mouth or injection into a vein,
these include pneumonia, cellulitis, urinary
tract infections, prostatitis, plague,
and certain types of infectious diarrhea. Other
uses, along with other medications, include treating multidrug resistant tuberculosis.[3] An eye
drop may be used for a superficial
bacterial infection of the eye and
an ear drop may
be used for otitis media when
a hole in the ear drum is
present.Ofloxacin is a broad-spectrum antibiotic that is active against both Gram-positive and Gram-negative bacteria.
It functions by inhibiting two bacterial type II topoisomerases, DNA
gyrase and topoisomerase IV. Topo IV is an enzyme necessary to separate (mostly in prokaryotes, in bacteria in particular) replicated DNA, thereby inhibiting bacterial cell division.
|
Sparfloxacin
https://en.wikipedia.org/wiki/Sparfloxacin
![]()
.Sparfloxacin is
a fluoroquinolone antibiotic used
in the treatment of bacterial infections. It has a controversial safety profile.Sparfloxacin, like other quinolones and fluoroquinolones,
are bactericidal drugs, actively killing bacteria. Quinolones inhibit the bacterial DNA gyrase or the topoisomerase
IV enzyme, thereby inhibiting DNA replication and transcription. Quinolones can enter cells easily and therefore are often used to treat intracellular pathogens such as Legionella pneumophila and Mycoplasma pneumoniae. For many gram-negative bacteria DNA gyrase is the target, whereas topoisomerase IV is the target for many gram-positive bacteria. Eukaryotic cells do not contain DNA gyrase or topoisomerase
IV.
|
| Tetracyclines |
Tigecycline
https://en.wikipedia.org/wiki/Tigecycline

Tigecycline is
an antibiotic for
a number of bacterial infections. It
is a glycylcycline administered
intravenously. It was developed in response to the growing rate of antibiotic resistant bacteria
such as Staphylococcus aureus, Acinetobacter
baumannii, and E.
coli. As
a tetracycline derivative antibiotic, its structural modifications has expanded its therapeutic activity to include Gram-positive and Gram-negative organisms, including those of multi-drug resistance.
Tigecycline is broad-spectrum antibiotic that acts as a protein synthesis inhibitor. It exhibits bacteriostatic activity by binding to the 30S ribosomal subunit of bacteria and thereby blocking the interaction of aminoacyl-tRNA with
the A site of the ribosome. In addition, tigecycline has demonstrated bactericidal activity against isolates of S. pneumoniae and L. pneumophila.
It is a third generation tetracycline derivative within a class called glycylcyclines which carry a N,N-dimethyglycylamido (DMG) moiety attached to the 9-position of tetracycline ring D. With structural modifications as a 9-DMG derivative of minocycline, tigecycline has been found to improve minimal inhibitory concentrations against Gram-negative and
Gram-positive organisms, when compared to tetracyclines.
|
| |
|
| Antidiabetic & pcos |
| Antidiabetic Agents |
Repaglinide
https://en.wikipedia.org/wiki/Repaglinide
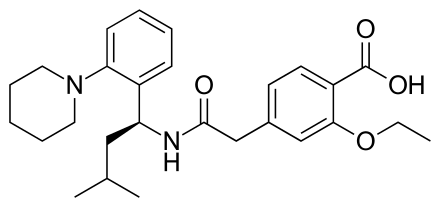
Repaglinide is
an antidiabetic drug in
the class of medications known as meglitinides,
and was invented in 1983. Repaglinide is an oral medication used in addition to diet and exercise for blood sugar control in type 2 diabetes mellitus. The
mechanism of action of repaglinide involves promoting insulin release from β-islet cells of the pancreasRepaglinide lowers blood glucose by stimulating the release of insulin from the beta islet cells of the pancreas.
It achieves this by closing ATP-dependent potassium
channels in the membrane of the beta cells. This depolarizes the beta cells, opening the cells' calcium
channels, and the resulting calcium influx induces insulin secretion.
|
Metformin HCl
https://en.wikipedia.org/wiki/Metformin

Metformin, marketed under the trade name Glucophage among
others, is the first-line medication
for the treatment of type 2 diabetes, particularly
in people who are overweight. It
is also used in the treatment of polycystic ovary syndrome. It
is not associated with weight gain.Metformin is the most widely used medication for diabetes taken by mouth. It is available as a generic medication. In 2017, it was the fourth-most commonly prescribed medication in the United States, with more than 78 million prescriptions.
The molecular mechanism of metformin is not completely understood. Multiple potential mechanisms of action have been proposed: inhibition of the mitochondrial respiratory chain (complex I), activation of AMP-activated protein kinase (AMPK), inhibition of glucagon-induced elevation of cyclic
adenosine monophosphate (cAMP) with reduced activation of protein kinase A (PKA), inhibition of mitochondrial glycerophosphate
dehydrogenase, and an effect on gut microbiota. Metformin also exerts an anorexiant effect in most people, decreasing caloric intake. Ultimately, it decreases gluconeogenesis (glucose
production) in the liver. It also has an insulin-sensitizing effect with multiple actions on tissues including the liver, skeletal muscle, endothelium, adipose tissue, and the ovary. The average patient with type 2 diabetes has three times the normal rate of gluconeogenesis; metformin treatment reduces this by over one-third. |
| Diabetic Neuropathy |
Epalrestat
https://en.wikipedia.org/wiki/Epalrestat

Epalrestat is
a carboxylic acid derivative[1] and
a noncompetitive and reversible aldose reductase inhibitor used
for the treatment of diabetic neuropathy,
which is one of the most common long-term complications in patients with diabetes mellitus.
It reduces the accumulation of intracellular sorbitol which is believed to be the cause of diabetic neuropathy, retinopathy and nephropathyIt has been proven in animal experiments that there is an improvement in sorbitol levels and Na+/K+ ATPase activity leading to improved nerve conduction velocity. Diabetic rats treated with epalrestat showed improvement in morphological abnormalities of nerves.
|
Tapentadol HCl
https://en.wikipedia.org/wiki/Tapentadol

Tapentadol, brand names Nucynta among others, is a centrally acting opioid analgesic of the benzenoid class with a dual mode of action as an agonist of the μ-opioid receptor and as a norepinephrine
reuptake inhibitor (NRI). Analgesia occurs within 32 minutes of oral administration, and lasts for 4–6 hours.
It is similar to tramadol in its dual mechanism of action; namely, its ability to activate the mu opioid receptor and inhibit the reuptake of norepinephrine. Unlike tramadol, it has only weak effects on the reuptake of serotonin and is a significantly more potent opioid with no known active metabolites Tapentadol is not a pro-drug and therefore does not rely on metabolism to produce its therapeutic effects; this makes it a useful moderate-potency analgesic option for patients who do not respond adequately to more commonly used opioids due to genetic disposition (poor metabolizers of CYP3A4 and CYP2D6),
as well as providing a more consistent dosage-response range among the patient population.
The potency of tapentadol is somewhere between that of tramadol and morphine, with an analgesic efficacy comparable to that of oxycodone despite a lower incidence of side effects. It is generally regarded as a weak-moderate strength opioid (a category shared by many better-known opioids such as hydrocodone and pethidine).
|
| DPP-4 Inhibitor (Gliptin) |
Alogliptin
https://en.wikipedia.org/wiki/Alogliptin

.Alogliptin (trade
name Nesina and Vipidia
is an oral anti-diabetic drug in
the DPP-4 inhibitor (gliptin)
class. Alogliptin does
not decrease the risk of heart attack and stroke. Like other members of the gliptin class, it causes little or no weight gain, exhibits relatively little risk of hypoglycemia, and has relatively modest glucose-lowering activity. Alogliptin and other gliptins are commonly used in combination with metformin in people whose diabetes cannot adequately be controlled with metformin alone.Alogliptin is a dipeptidyl peptidase-4 inhibitor that decreases blood
sugar similar to the other.
|
Teneligliptin
https://en.wikipedia.org/wiki/Teneligliptin

Teneligliptin (INN;
trade name Tenelia) is a pharmaceutical drug for the
treatment of type 2 diabetes mellitus.
It belongs to the class of anti-diabetic drugs known as dipeptidyl peptidase-4 inhibitors or
"gliptins".
|
| Gestational Diabetes |
Inositol
https://en.wikipedia.org/wiki/Inositol
Inositol,
or more precisely myo-inositol, is a carbocyclic sugar that
is abundant in brain and other mammalian tissues, mediates cell signal transduction in response to a variety of hormones, neurotransmitters and
growth factors and participates in osmoregulation. It
is a sugar alcohol with
half the sweetness of sucrose (table
sugar). It is made naturally in humans from glucose.
A human kidney makes about two grams per day. Other tissues synthesize it too, and the highest concentration is in the brain where it plays an important role making other neurotransmitters and
some steroid hormones bind
to their receptors. Since
about 2008 myo-inositol gained importance in the management of polycystic
ovary syndrome (PCOS)
due to its efficacy, safety profile and worldwide availability.
|
| PCOS |
D-Chiro-Inositol
https://en.wikipedia.org/wiki/1D-chiro-Inositol

1D-chiro-Inositol (formerly D-chiro-inositol,
commonly abbreviated DCI) is a member of a family of related substances
often referred to collectively as "inositol",
although that term encompasses several isomers of
questionable biological relevance, including 1L-chiro-inositol. myo-Inositol
is converted into DCI by an insulin dependent NAD/NADH epimerase enzyme.It is known to be an important secondary messenger in insulin signal
transduction. DCI accelerates the dephosphorylation of glycogen synthase and pyruvate dehydrogenase, rate limiting enzymes of non-oxidative and oxidative glucose disposal. DCI may act to bypass defective normal epimerization of myo-inositol
to DCI associated with insulin resistance and at least partially restore insulin sensitivity and glucose disposal.
|
| SGLT-2 Inhibitor (Gliflozin) |
Canagliflozin
https://en.wikipedia.org/wiki/Canagliflozin

Canagliflozin, sold under the brand name Invokana among
others, is a medication used to treat type 2 diabetes. It
is a third-line medication to metformin. It
is used together with exercise and diet. It is not recommended in type 1 diabetes. It
is taken by mouth.
Canagliflozin is an inhibitor of subtype 2 sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of renal glucose reabsorption (the remaining 10% is done by SGLT1). Blocking this transporter causes up to 119 grams of blood glucose
per day to be eliminated through the urine, corresponding to 476 kilocalories. Additional water is eliminated by osmotic diuresis, resulting in a lowering of blood pressure.
This mechanism is associated with a low risk of hypoglycaemia (too low blood glucose) compared to other types of anti-diabetic drugs such as sulfonylurea derivatives and insulin.
|
| |
|
| Cardiovascular system |
| Alpha/Beta Adrenoceptor Agonist |
Carvedilol HCl / Phosphate
https://en.wikipedia.org/wiki/Carvedilol

Carvedilol,
sold under the brand name Coreg among
others, is a medication used to treat high blood pressure, congestive
heart failure (CHF), and left
ventricular dysfunction in
people who are otherwise stable. For
high blood pressure, it is generally a second-line treatment. It
is taken by mouth.[
Carvedilol is both a non-selective beta adrenergic receptor blocker (β1, β2) and an alpha adrenergic receptor blocker (α1). The S(-) enantiomer accounts for the beta blocking activity whereas the S(-) and R(+) enantiomer have alpha blocking activity.
Carvedilol reversibly binds to beta adrenergic receptors on cardiac myocytes. Inhibition of these receptors prevents a response to the sympathetic nervous system, leading to decreased heart rate and contractility. This action is beneficial in heart failure patients where the sympathetic nervous system is activated as a compensatory mechanism.
Carvedilol blockade of α1 receptors causes vasodilation of blood vessels. This inhibition leads to decreased peripheral vascular resistance and an antihypertensive effect. There is no reflex tachycardia response due to carvedilol blockade of β1 receptors on the heart.
|
Eplerenone
https://en.wikipedia.org/wiki/Eplerenone

Eplerenone,
sold under the brand name Inspra, is a steroidal antimineralocorticoid of
the spirolactone group
that is used as an adjunct in the management of chronic heart failure and high
blood pressure, particularly for patients with resistant hypertension due to elevated aldosterone. Classed as a selective
aldosterone receptor antagonist (SARA), it
is similar to the diuretic spironolactone,
though it is much more selective for
the mineralocorticoid receptor in
comparison (i.e., does not possess any antiandrogen, progestogen, glucocorticoid,
or estrogenic effects),
and is specifically marketed for educing cardiovascular risk
in patients following myocardial infarction.
Eplerenone is a potassium-sparing diuretic,
meaning that it helps the body get rid of water but still keep potassium.
|
Fenoterol HBr
https://en.wikipedia.org/wiki/Fenoterol

Fenoterol is
a β adrenoreceptor agonist.
It is classed as sympathomimetic β2 agonist
and an inhaled bronchodilator asthma medication.5-(1-Hydroxy-2-{[2-(4-hydroxyphenyl)-1-methylethyl]amino}ethyl)benzene-1,3-diol is a molecule with two different stereogenic centers. Thus, four stereoisomers may exist, the (R,R)-,
(R,S)-, (S,R)-
and (S,S)-stereoisomers (see the figure below). Fenoterol is a racemate of
the (R,R)- and the (S,S)-enantiomers.
This racemate is 9 to 20 times more effective, as compared to the racemate of the (R,S)- and (S,R)-enantiomers. |
Icatibant Acetate
https://en.wikipedia.org/wiki/Icatibant

Icatibant, sold under the brand name Firazyr, is a medication for the symptomatic treatment of acute attacks of hereditary angioedema (HAE) in adults with C1-esterase-inhibitor deficiency.It is not effective in angioedema caused by medication from the ACE inhibitor class.
It is a peptidomimetic consisting of ten amino acids, which is a selective and specific antagonist of bradykinin B2 receptors. It cost about US$8,000 to 9,000 per dose as of 2014.
|
Imiquimod
https://en.wikipedia.org/wiki/Imiquimod

Imiquimod (INN)
is a prescription medication that acts as an immune response modifier that
is used to treat genital warts,
superficial basal cell carcinoma,
and actinic keratosis.
miquimod yields profound antitumoral activity by acting on several immunological levels synergistically.[9] Imiquimod stimulates the innate immune system by activating toll-like receptor 7 (TLR7), commonly involved in pathogen recognition.Cells activated by imiquimod via TLR-7 secrete cytokines (primarily interferon-α (IFN-α), interleukin-6 (IL-6),
and tumor necrosis factor-α (TNF-α)).There is evidence that imiquimod, when applied to skin, can lead to the activation of Langerhans cells, which subsequently migrate to local lymph nodes to activate the adaptive immune system. Other cell types activated by imiquimod include natural
killer cells, macrophages and B-lymphocytes.
Imiquimod exerts its effect by increasing levels of the opioid growth factor receptor (OGFr). In experiments, blocking OGFr function with siRNA technology resulted in loss of any antiproliferative effect of imiquimod.
|
Isoprenaline HCl
https://en.wikipedia.org/wiki/Isoprenaline
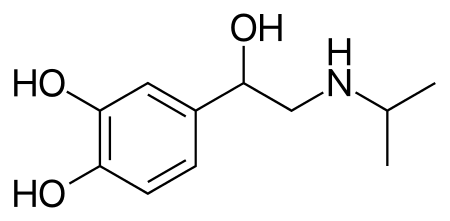
Isoprenaline,
or isoproterenol, is a medication used
for the treatment of bradycardia (slow
heart rate) heart block,
and rarely for asthma.
It is a non-selective β adrenoreceptor agonist that
is the isopropylamine analog of epinephrine (adrenaline).Isoprenaline
is a β1 and β2 adrenoreceptor
agonist and has almost no activity on alpha
adrenergic receptors. Its agonist effects at TAAR1 provide
it with a pharmacodynamic effects that resemble those of the endogenous trace amines, like tyramine.The isopropylamine group
in isoprenaline makes it selective for β receptors. The free catechol hydroxyl groups
keep it susceptible to enzymatic metabolism.
|
Levosalbutamol HCl / Sulphate / Tartrate
https://en.wikipedia.org/wiki/Levosalbutamol

Levosalbutamol, also known as levalbuterol, is a short-acting β2 adrenergic receptor agonist used in the treatment of asthma and chronic obstructive pulmonary disease (COPD). Evidence does not
show that levosalbutamol works better than salbutamol; thus there may not be sufficient justification for prescribing it.
The drug is the (R)-(−)-enantiomer of its prototype drug salbutamol.
Activation of β2 adrenergic receptors on airway smooth muscle leads to the activation of adenylate cyclase and to an increase in the intracellular concentration of 3',5'-cyclic adenosine monophosphate (cyclic AMP). The increase in cyclic AMP is associated with
the activation of protein kinase A, which in turn, inhibits the phosphorylation of myosin and lowers intracellular ionic calcium concentrations, resulting in muscle relaxation.
Levosalbutamol relaxes the smooth muscles of all airways, from the trachea to the terminal bronchioles. Increased cyclic AMP concentrations are also associated with the inhibition of the release of mediators from mast cells in the airways. Levosalbutamol acts as a functional agonist that relaxes the airway irrespective of the spasmogen involved, thereby protecting against all bronchoconstrictor challenges.
While it is recognized that β2 adrenergic receptors are the predominant receptors on bronchial smooth muscle, data indicate that there are beta receptors in the human heart, 10–50% of which are β2 adrenergic receptors. The precise function of these receptors has not been established. However, all β adrenergic agonist drugs can produce a significant cardiovascular effect in some patients, as measured by pulse rate, blood pressure, and restlessness symptoms, and/or electrocardiographic (ECG).
|
Macitentan
https://en.wikipedia.org/wiki/Macitentan

Macitentan (trade
name Opsumit) is an endothelin
receptor antagonist (ERA)
approved for the treatment of pulmonary arterial hypertension (PAH). The
other two ERAs marketed as of 2014 are bosentan and ambrisentan. Macitentan
is a dual ERA, meaning that it acts as an antagonist of two endothelin (ET)
receptor subtypes, ETA and ETB. However,
macitentan has a 50-fold increased selectivity for the ETA subtype compared to the ETB subtype..Endothelin (ET) is an extremely potent blood vessel constricting substance that is secreted by endothelial cells. In
the lungs, the most common ET form released is ET-1. ET-1
release can occur through both constitutive and non-constitutive pathways. Upon
release, ET-1 can bind to the ET receptors that are expressed on arterial smooth muscle cells and fibroblasts in the lungs. ET
receptors are G protein coupled receptors and, when activated, lead to an increase in intracellular calcium levels via the Gαq signaling
pathway. The rise in
intracellular calcium leads to contraction of the arterial smooth muscle, as well as vascular remodelling due to cell proliferation. Prolonged
constriction and fibrosis are factors in the pathogenesis of PAH.
|
Ractopamine HCl
https://en.wikipedia.org/wiki/Ractopamine

Ractopamine is an animal feed additive used to promote leanness and increase food conversion efficiency in livestock in some countries, but banned in others. Pharmacologically, it is a phenol-based TAAR1 agonist and β adrenoreceptor agonist that stimulates β1 and β2 adrenergic
receptors.It is most commonly administered to animals for meat production as ractopamine hydrochloride.It is the active ingredient in products marketed in the US as Paylean for swine, Optaflexx for cattle, and Topmax for turkeys.It was developed by Elanco Animal Health, a division of Eli Lilly and Company.
As of 2014 the use of ractopamine was banned in 160 countries, including the European Union, mainland China and Russia, while 27 other countries, such as Japan, the United
States, South Korea, and New Zealand have deemed meat from livestock fed ractopamine safe for human consumption.
Commercial ractopamine is a mixture of all four possible stereoisomers.It is also a positional isomer of dobutamine, a related drug.
|
Silodosin
https://en.wikipedia.org/wiki/Silodosin

Silodosin (trade
names Urief in
Japan, Rapaflo in
the US, and Silodyx in
Europe) is a medication for the symptomatic treatment of benign
prostatic hyperplasia (BPH).
It acts as an α1-adrenoceptor antagonist with
high uroselectivity (selectivity for the prostate).Silodosin
has high affinity for the α1A adrenergic receptor
in the prostate, the bladder, and the prostatic
urethra. By this mechanism it
relaxes the smooth muscle in these organs, easing urinary flow and other symptoms of BPH.
|
Tizanidine HCl
https://en.wikipedia.org/wiki/Tizanidine

Tizanidine,
sold under the brand name Zanaflex among
others, is a medication that is used to treat muscle spasticity due
to spinal cord injury or multiple
sclerosis. Effectiveness
appears similar to baclofen or diazepam.It
is taken by mouth.[Tizanidine
is an α2 receptor agonist
closely related to clonidine. It
has approximately one tenth to one fifteenth of the pressure lowering effect of clonidine. The relation between the α2 receptor
agonism and the spasmolytic action is still not fully understood.
|
Tulobuterol Base / HCl
https://en.wikipedia.org/wiki/Tulobuterol

Tulobuterol (INN) is a long-acting beta2-adrenergic receptor agonist, marketed in Japan as a transdermal patch under the name Hokunalin tape (ホクナリンテープ).
Currently, it is only legal in 7 countries: Japan, Germany, China, South Korea, Bangladesh, Pakistan, and Venezuela. It is available in India also.
|
| Anti-Anginal Drug |
Amlodipine
https://en.wikipedia.org/wiki/Amlodipine

Amlodipine,
sold under the brand name Norvasc among
others, is a medication used to treat high blood pressure and coronary
artery disease.[6] While
not typically recommended in heart failure,
amlodipine may be used if other medications are not sufficient for treating high blood pressure or heart-related chest pain. It
is taken by mouth and has an effect that lasts for at least a day.In 2017, it was the fifth most commonly prescribed medication in the United States, with more than 72 million prescriptions.
Amlodipine is an angioselective calcium channel blocker and inhibits the movement of calcium ions into vascular smooth muscle cells and cardiac muscle cells which inhibits the contraction of cardiac muscle and vascular smooth muscle cells. Amlodipine inhibits calcium ion influx across cell membranes, with a greater effect on vascular smooth muscle cells. This causes vasodilation and a
reduction in peripheral vascular resistance, thus lowering blood pressure. Its effects on cardiac muscle also prevent excessive constriction in the coronary arteries.
Negative inotropic effects can be detected in vitro, but such effects have not been seen in intact animals at therapeutic doses. Among the two stereoisomers [R(+), S(–)], the (–) isomer has been reported to be more active than the (+) isomer.[36] Serum
calcium concentration is not affected by amlodipine. And it specifically inhibits the currents of L-type Cav1.3 channels in the zona glomerulosa of the adrenal
gland.
|
Isosorbide 5 MonoNitrate
https://en.wikipedia.org/wiki/Isosorbide_mononitrate

Isosorbide mononitrate,
sold under many brand names, is a medication used for heart-related chest pain (angina), heart
failure, and esophageal
spasms. It
can be used both to treat and to prevent eart-related chest pain; however, it is generally less preferred than beta
blockers or calcium
channel blockers. It
is taken by mouth.
|
Isosorbide Dinitrate
https://en.wikipedia.org/wiki/Isosorbide_dinitrate

Isosorbide dinitrate (ISDN)
is a medication used for heart failure, esophageal
spasms, and to treat and prevent chest
pain from not enough blood flow to the heart. It
has been found to be particularly useful in heart failure due to systolic dysfunction together
with hydralazine in black
people. It
is taken by mouth or
under the tongue.
Similar to other nitrites and organic nitrates, isosorbide dinitrate is converted to nitric oxide (NO), an active intermediate compound which activates the enzyme guanylate cyclase (atrial natriuretic peptide receptor A). This stimulates the synthesis of cyclic guanosine 3',5'-monophosphate (cGMP) which then activates a series of protein kinase-dependent phosphorylations in the smooth muscle cells, eventually resulting in the dephosphorylation of the myosin light chain of the smooth muscle fiber. The subsequent sequestration of calcium ions results in the relaxation of the smooth muscle cells and vasodilation.
|
Ivabradine
https://en.wikipedia.org/wiki/Ivabradine
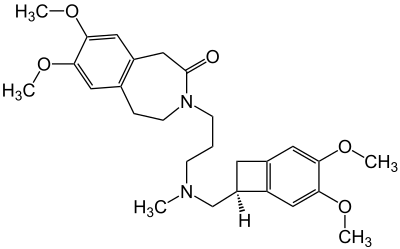
Ivabradine, marketed under trade names including Coralan, Corlentor, Procoralan, Coraxan, and many more,is a medication used for the symptomatic management of stable heart-related chest pain and heart failure not fully managed by beta
blockers.
Ivabradine acts by reducing the heart rate via specific inhibition of the pacemaker current, a mechanism different from that of beta blockers and calcium channel blockers, two commonly prescribed antianginal drugs.
Ivabradine is a cardiotonic agent.
Ivabradine acts on the If (f is for "funny", so called because it had unusual properties compared with other current systems known at the time of its discovery) ion current, which is highly expressed in the sinoatrial node. If is a mixed Na+–K+ inward
current activated by hyperpolarization and modulated by the autonomic nervous system. It is one of the most important ionic currents for regulating pacemaker activity in the sinoatrial (SA) node. Ivabradine selectively inhibits the pacemaker If current in a dose-dependent manner. Blocking this channel reduces cardiac pacemaker activity, selectively slowing the heart rate and allowing more time for blood to flow to the myocardium. This is in contrast to other commonly used rate-reducing medications, such as beta-blockers and calcium channel blockers, which not only reduce heart rate, but also the cardiac contractility. Given the selective decrease in rate without loss of
contractility, ivabradine may prove efficacious for treatment of congestive heart failure with reduced ejection fraction.
Ivabradine binds to HCN4 receptors (Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4), utilizing Y506, F509 and I510 residues.
|
Nifedipine
https://en.wikipedia.org/wiki/Nifedipine

Nifedipine,
sold under the brand name Adalat among
others, is a medication used to manage angina, high
blood pressure, Raynaud's
phenomenon, and premature
labor. It
is one of the treatments of choice for Prinzmetal angina. It
may be used to treat severe high blood pressure in pregnancy.Its
use in preterm labor may allow more time for steroids to
improve the baby's lung function and provide time for transfer of the mother to a well qualified medical facility before delivery. It
is a calcium channel blocker of
the dihydropyridine type. Nifedipine
is taken by mouth and comes in fast and slow release formulations..
Nifedipine is a calcium channel blocker. Although nifedipine and other dihydropyridines are commonly regarded as specific to the L-type calcium channel, they also possess nonspecific activity towards other voltage-dependent
calcium channels.
Nifedipine has additionally been found to act as an antagonist of the mineralocorticoid receptor, or as an antimineralocorticoid
.
|
Pazopanib
https://en.wikipedia.org/wiki/Pazopanib

Pazopanib (trade
name Votrient) is an anti-cancer drug. It is a potent and
selective multi-targeted receptor tyrosine kinase inhibitor that
blocks tumour growth and inhibits angiogenesis.
It has been approved for renal cell carcinoma and soft
tissue sarcoma by numerous
regulatory administrations worldwide..Pazopanib is a multiple kinase inhibitor that limits tumor growth by targeting
angiogenesis via inhibition
of enzymes including vascular endothelial growth factor receptor (VEGFR),
platelet-derived growth factor receptor (PDGFR), c-KIT and FGFR.
|
| Antianemic Agent |
Ferrous Bisglycinate
https://en.wikipedia.org/wiki/List_of_combined_sex-hormonal_preparations
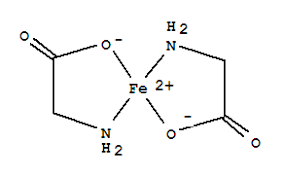
.Glycine (symbol Gly or G; ) is
an amino acid that
has a single hydrogen atom
as its side chain.
It is the simplest amino acid (since carbamic acid is
unstable), with the chemical formula NH2‐CH2‐COOH.
Glycine is one of the proteinogenic amino acids.
It is encoded by
all the codons starting
with GG (GGU, GGC, GGA, GGG). Glycine is not widely used in foods for its nutrional value, except in infusions. Instead glycine's role in food chemistry is as a flavorant. It is mildly sweet, and it counters the aftertaste of saccharine. It also has preservative properties, perhaps owing to its complexation to metal ions. Metal glycinate complexes, e.g. copper(II)
glycinate are used as supplements for animal feeds..
|
| Antiarrhythmic Drugs |
Amiodarone HCl
https://en.wikipedia.org/wiki/Tramadol

Tramadol,
sold under the brand name Ultram among
others, is an opioid pain
medication used to treat
moderate to moderately severe pain. When
taken by mouth in an immediate-release formulation, the onset of pain relief usually begins within an hour. It
is also available by injection. It
may be sold in combination with paracetamol (acetaminophen)
or as longer-acting formulations.Tramadol induces analgesic effects through a variety of different targets on the noradrenergic
system, serotoninergic
system and opioid receptors system. Tramadol exists as a racemic mixture, the positive enantiomer inhibits
serotonin reuptake while the negative enantiomer inhibits noradrenaline re-uptake, by binding to and blocking the transporters. Tramadol has also been shown to act as a serotonin releasing agent. Both enantiomers of tramadol are agonists of the μ-opioid receptor and its M1 metabolite, O-demethylate,
is also a μ-opioid receptor agonist but is 6 times more potent than tramadol itself. All these effects work synergistically to induce analgesia..
|
Diltiazem
https://en.wikipedia.org/wiki/Diltiazem

Diltiazem, sold under the brand name Cardizem among
others, is a calcium channel blocker medication
used to treat high blood pressure, angina,
and certain heart arrhythmias. It
may also be used in hyperthyroidism if beta
blockers cannot be used. It
is taken by mouth or injection
into a vein.When given by injection, effects typically begin within a few minutes and last a few hours.Diltiazem, also known as (2S,3S)-3-acetoxy-5-[2-(dimethylamino)ethyl]-2,3-dihydro-2-(4-methoxyphenyl)-1,5-benzothiazepin-4(5H)-one hydrochlorid has a vasodilating activity attributed to the (2S,3S)-isomer. Diltiazem is a potent vasodilator, increasing blood flow and variably decreasing the heart
rate via strong depression of A-V node conduction. Its pharmacological activity is somewhat similar to verapamil,
another nondihydropyridine (non-DHP) calcium channel blocker. Chemically, it is based upon a 1,4-thiazepine ring,
making it a benzothiazepine-type calcium channel blocker..
|
Dronedarone
https://en.wikipedia.org/wiki/Dronedarone

Dronedarone (development
codename SR33589 and
marketed as Multaq) is a drug by Sanofi-Aventis,
mainly for the indication of cardiac arrhythmias.
It was approved by the FDA on July 2, 2009. It was recommended as an alternative to amiodarone for
the treatment of atrial fibrillation and atrial
flutter in people whose
hearts have either returned to normal rhythm or who undergo drug therapy or electric shock treatment i.e.
direct current cardioversion (DCCV)
to maintain normal rhythm..Dronedarone has been termed a “multichannel blocker” however it is unclear which channel(s) play a pivotal role in its success. Thus,
dronedarone's actions at the cellular level are controversial with most studies suggesting an inhibition in multiple outward potassium currents including rapid delayed rectifier, slow delayed rectifier and ACh-activated inward rectifier. It
is also believed to reduce inward rapid Na current and L-type Ca channels.
|
Lidocaine (Lignocaine)
https://en.wikipedia.org/wiki/Lidocaine
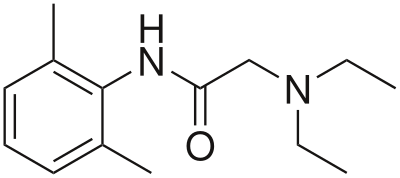
.Lidocaine,
also known as lignocaine, is a medication used to numb
tissue in a specific area (local
anesthetic) It is also
used to treat ventricular tachycardia and
to perform nerve blocks. Lidocaine
mixed with a small amount of adrenaline (epinephrine)
is available to allow larger doses for numbing, to decrease bleeding, and to make the numbing effect last longer.[4] When
used as an injectable, lidocaine typically begins working within four minutes and lasts for half an hour to three hours. Lidocaine
mixtures may also be applied directly to the skin or mucous membranes to
numb the area.
Lidocaine alters signal conduction in neurons by prolonging the inactivation of the fast voltage-gated Na+ channels in the neuronal cell membrane responsible for action potential propagation. With sufficient blockage, the
voltage-gated sodium channels will not open and an action potential will not be generated. Careful titration allows for a high degree of selectivity in the blockage of sensory neurons, whereas higher concentrations also affect other types of neurons.
The same principle applies for this drug's actions in the heart. Blocking sodium channels in the conduction system, as well as the muscle cells of the heart, raises the depolarization threshold, making the heart less likely to initiate or conduct early action potentials that may cause an arrhythmia.
|
| Anticoagulants |
Dabigatran Etexilate
https://en.wikipedia.org/wiki/Dabigatran

.Dabigatran,
sold under the brand name Pradaxa among
others, is an anticoagulant used
to treat and prevent blood clots and
to prevent stroke in
people with atrial fibrillation. Specifically
it is used to prevent blood clots following hip or knee
replacement and in those
with a history of prior clots. It
is used as an alternative to warfarin and
does not require monitoring by blood tests. It
is taken by mouth.Dabigatran reversibly binds to the active site on the thrombin molecule, preventing thrombin-mediated activation of coagulation
factors. Furthermore, dabigatran can inactivate thrombin even when thrombin is fibrin-bound; it reduces thrombin-mediated inhibition of fibrinolysis and,
therefore, may enhance fibrinolysis.
|
Enoxaparin
https://en.wikipedia.org/wiki/Enoxaparin_sodium

Enoxaparin sodium is
an anticoagulant medication
(blood thinner). It is
used to treat and prevent deep vein thrombosis (DVT)
and pulmonary embolism (PE)
including during pregnancy and
following certain types of surgery. It
is also used in those with acute coronary syndrome (ACS)
and heart attacks. It
is given by injection just under the skin or into
a vein. Other
uses include inside kidney dialysis machines.Enoxaparin
binds to and potentiates antithrombin (a
circulating anticoagulant) to form a complex that irreversibly inactivates clotting factor
Xa. It has less activity against factor
IIa (thrombin) compared to unfractionated heparin (UFH) due to its low molecular weight.
|
Warfarin Sodium Clathrate
https://en.wikipedia.org/wiki/Warfarin

Warfarin, sold under the brand name Coumadin among
others,is a medication that is used as an anticoagulant (blood
thinner). It
is commonly used to treat blood clots such
as deep vein thrombosis and pulmonary
embolism, and to prevent stroke in
people who have atrial fibrillation, valvular
heart disease or artificial
heart valves. Less
commonly it is used following ST-segment elevation myocardial infarction (STEMI)
and orthopedic surgery.
Warfarin decreases blood clotting by blocking an enzyme called vitamin K epoxide reductase that reactivates vitamin K1. Without sufficient active vitamin K1, clotting
factors II, VII, IX, and X have decreased clotting ability. The anticlotting protein C and protein S are also inhibited, but to a lesser degree. A few days are required for full effect to occur, and these effects can last for up to five days.
Warfarin first came into commercial use in 1948 as a rat poison. In 1954 it was approved for medical use in the United States.
|
| Antihypertensives |
Cilnidipine
https://en.wikipedia.org/wiki/Cilnidipine

Cilnidipine is a calcium channel blocker. Cilnidipine is approved for use in Japan, China, India, Korea, and some European countries to treat hypertension.
It is a calcium antagonist accompanied with L-type and N-type calcium channel blocking functions. Unlike other calcium antagonists, cilnidipine can act on the N-type calcium channel in addition to acting on the L-type
calcium channel.
Cilnidipine decreases blood pressure and is used to treat hypertension and its comorbidities.
Due to its blocking action at the N-type
and L-type calcium channel,
cilnidipine dilates both arterioles and venules,
reducing the pressure in the capillary bed.
Cilnidipine is vasoselective and has a weak direct dromotropic effect, a strong vasodepressor effect, and an arrhythmia-inhibiting effect. |
Irbesartan
https://en.wikipedia.org/wiki/Irbesartan

Irbesartan,
sold under the brand name Avapro among
others, is a medication used to treat high blood pressure, heart
failure, and diabetic
kidney disease. It
is a reasonable initial treatment for high blood pressure.[2] It
is taken by mouth.[2] Versions
are available as the combination irbesartan/hydrochlorothiazide.As
with all angiotensin II receptor antagonists, irbesartan is used for the treatment of hypertension. It may also delay progression of diabetic nephropathy and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes, hypertension
and microalbuminuria (>30 mg/24 h) or proteinuria (>900 mg/24 h)..
|
Losartan Potassium
https://en.wikipedia.org/wiki/Losartan
All of the physiological effects of angiotensin II, including release of aldosterone,
are antagonized in the presence of losartan. Reduction in blood pressure occurs independently of the status of the renin–angiotensin system.
As a result of losartan dosing, plasma renin activity
increases due to removal of the angiotensin II feedback. Renin is released from the kidneys when there is reduced renal arterial pressure, sympathetic activation, or increased sodium delivery to the distal renal tubule. Renin then acts by converting angiotensinogen to angiotensin I; angiotensin converting enzyme (ACE) converts angiotensin I to angiotensin II; angiotensin II causes vasoconstriction and aldosterone release. Aldosterone
serves to retain sodium from the distal renal tubule. Sodium retention ultimately results in increased blood pressure. Therefore, the use of angiotensin II receptor antagonists like losartan result in blocking the downstream effect of renin, angiotensin II, and ultimately decreasing blood pressure.
|
Moexipril
https://en.wikipedia.org/wiki/Moexipril

Moexipril an angiotensin converting enzyme inhibitor (ACE inhibitor) used for the treatment of hypertension and congestive heart failure. Moexipril can be administered alone or with other antihypertensives or diuretics.
It works by inhibiting the conversion of angiotensin I to angiotensin II.As
an ACE inhibitor, moexipril causes a decrease in ACE. This blocks the conversion of angiotensin I to angiotensin II. Blockage of angiotensin II limits hypertension within the vasculature. Additionally, moexipril has been found to possess cardioprotective properties.
|
Sodium Nitroprusside
https://en.wikipedia.org/wiki/Sodium_nitroprusside

Sodium nitroprusside (SNP),
sold under the brand name Nitropress among
others, is a medication used to lower blood pressure. This
may be done if the blood pressure is very high and resulting in symptoms,
in certain types of heart failure,
and during surgery to decrease bleeding. It
is used by continuous injection into a vein.Onset
is typically immediate and effects last for up to ten minutes..
As a result of its breakdown to nitric oxide (NO), sodium nitroprusside has potent vasodilating effects on arterioles and venules (veins more than arteries) but this selectivity is much less marked than that of nitroglycerin.
Sodium nitroprusside breaks down in circulation to release nitric oxide (NO). It does this by binding to oxyhaemoglobin to release cyanide, methaemoglobin and nitric oxide. NO activates guanylate cyclase in vascular smooth muscle and increases intracellular production of cGMP. cGMP activates protein
kinase G which activates phosphatases which inactivate myosin light chains.[28] Myosin light chains are involved in muscle contraction. The end result is vascular smooth muscle relaxation, which allow vessels to dilate.[This
mechanism is similar to that of phosphodiesterase 5 (PDE5) inhibitors such as sildenafil (Viagra) and tadalafil (Cialis), which elevate cGMP concentration by inhibiting its degradation by PDE5.
|
Atorvastatin Calcium
https://en.wikipedia.org/wiki/Atorvastatin

Atorvastatin, sold under the
brand name Lipitor among
others, is a statin medication
used to prevent cardiovascular disease in
those at high risk and treat abnormal lipid levels. For
the prevention
of cardiovascular
disease, statins are a first-line treatment.[As
with other statins, atorvastatin is a competitive inhibitor of HMG-CoA
reductase. Unlike most others, however, it is a completely synthetic compound.
HMG-CoA reductase catalyzes the reduction of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA)
to mevalonate,
which is the rate-limiting step in
hepatic cholesterol biosynthesis.
Inhibition of the enzyme decreases de novo cholesterol
synthesis, increasing expression of low-density lipoprotein receptors (LDL receptors)
on hepatocytes.
This increases LDL uptake by the hepatocytes, decreasing the amount of LDL-cholesterol in the blood. Like other statins, atorvastatin also reduces blood levels of triglycerides and
slightly increases levels of HDL-cholesterol. |
| Antiparkinsonian Drugs |
Levodopa
https://en.wikipedia.org/wiki/L-DOPA

l-DOPA
crosses the protective blood-brain barrier,
whereas dopamine itself
cannot. Thus, l-DOPA
is used to increase dopamine concentrations in the treatment of Parkinson's disease and dopamine-responsive
dystonia. Once l-DOPA
has entered the central nervous system,
it is converted into dopamine by the enzyme aromatic l-amino
acid decarboxylase, also known as DOPA
decarboxylase. Pyridoxal
phosphate (vitamin B6)
is a required cofactor in
this reaction,
and may occasionally be administered along with l-DOPA,
usually in the form of pyridoxine.Herbal
extracts containing l-DOPA are available; high-yielding sources
include Mucuna pruriens (velvet bean), and Vicia
faba (broad bean), while other sources include the genera Phanera, Piliostigma, Cassia, Canavalia,
and Dalbergia.
Rosuvastatin Calcium
https://en.wikipedia.org/wiki/Rosuvastatin

Rosuvastatin, sold under the trade name Crestor among
others, is a statin medication,
used to prevent cardiovascular disease in
those at high risk and treat abnormal lipids. It
is recommended to be used together with dietary changes, exercise, and weight loss. It is taken by mouth.
Rosuvastatin is a competitive inhibitor of the enzyme HMG-CoA reductase, having a mechanism of action similar to that of other statins.
Putative beneficial effects of rosuvastatin therapy on chronic heart failure may be negated by increases in collagen turnover markers as well as a reduction in plasma coenzyme Q10 levels in patients with chronic heart failure.
|
| |
|
| Antiplatelet Drugs |
Clopidogrel
https://en.wikipedia.org/wiki/Clopidogrel

Clopidogrel,
sold under the trade name Plavix among
others, is an antiplatelet
medication used to reduce
the risk of heart disease and stroke in
those at high risk. It
is also used together with aspirin in heart
attacks and following the
placement of a coronary artery stent (dual
antiplatelet therapy). It
is taken by mouth.[4] Onset
of effects is about two hours and lasts for five days.Clopidogrel is a prodrug, which is activated in two steps, first by CYP2C19, CYP1A2 and CYP2B6, then by CYP2C19, CYP2C9, CYP2B6 and CYP3A. The active metabolite then specifically and irreversibly inhibits the P2Y12 subtype
of ADP receptor, which is important in activation of platelets and eventual cross-linking by the protein fibrin. Platelet
inhibition can be demonstrated two hours after a single dose of oral clopidogrel, but the onset of action is slow, so a loading dose of either 600 or 300 mg is administered when a rapid effect is needed.
|
| Attention Deficit Hyperactivity Disorder (ADHD) |
Clonidine HCl
https://en.wikipedia.org/wiki/Clonidine

Clonidine,
sold as the brand name Catapres among
others, is a medication used to treat high blood pressure, attention
deficit hyperactivity disorder, drug
withdrawal (alcohol, opioids,
or smoking), menopausal
flushing, diarrhea,
and certain pain conditions.[7] It
is used by mouth, by injection, or as a skin patch.
Clonidine crosses the blood-brain barrier.
Clonidine treats high blood pressure by stimulating α2 receptors in the brain stem, which decreases peripheral vascular resistance, lowering blood pressure. It has specificity towards the presynaptic α2 receptors
in the vasomotor center in the brainstem. This binding has a sympatholytic effect, suppresses release of norepinephrine, ATP,
renin, and neuropeptide Y which if released would increase vascular resistance.
|
| Beta Blockers |
Atenolol
https://en.wikipedia.org/wiki/Atenolol

.Atenolol is
a beta blocker medication
primarily used to treat high blood pressure and heart-associated
chest pain.Atenolol,
however, does not seem to improve mortality in those with high blood pressure. Other
uses include the prevention of migraines and
treatment of certain irregular heart beats.
Atenolol is used for a number of conditions including hypertension, angina, long QT syndrome, acute myocardial infarction, supraventricular
tachycardia, ventricular tachycardia, and the symptoms of alcohol withdrawal.
The role for β-blockers in general in hypertension was downgraded in June 2006 in the United Kingdom, and later in the United States, as they are less appropriate than other agents such as ACE inhibitors, calcium channel blockers, thiazide diuretics and angiotensin
receptor blockers, particularly in the elderly.
|
Labetalol
https://en.wikipedia.org/wiki/Labetalol
Labetalol is
a medication used to treat high blood pressure and
in long term management of angina. This
includes essential hypertension, hypertensive emergencies,
and hypertension of pregnancy. In
essential hypertension it is generally less preferred than a number of other blood pressure medications.
Labetalol's dual alpha and beta adrenergic antagonism has different physiological effects in short- and long-term situations. In short-term, acute situations, labetalol decreases blood pressure by decreasing systemic vascular resistance with little effect on stroke
volume, heart rate and cardiac output.During long-term use, labetalol can reduce heart rate during exercise while maintaining cardiac output by an increase in stroke volume.
Labetalol is a dual alpha (α1) and beta (β1/β2) adrenergic receptor blocker and competes with other Catecholamines for binding to these sites. Its action on these receptors are potent and reversible Labetalol is highly selective for postsynaptic alpha1- adrenergic, and
non-selective for beta-adrenergic receptors. It is about equipotent in blocking beta1- and beta2- receptors.
|
Metoprolol
https://en.wikipedia.org/wiki/Metoprolol

Metoprolol,
marketed under the tradename Lopressor among
others, is a selective β1 receptor blocker medication. It
is used to treat high blood pressure, chest
pain due to poor blood flow to the heart, and a number of conditions involving an abnormally
fast heart rate. It
is also used to prevent further heart problems after myocardial infarction and
to prevent headaches in those with migraines.Metoprolol
blocks β1 adrenergic receptors in heart
muscle cells, thereby decreasing the slope of phase 4 in the
nodal action potential (reducing Na+ uptake) and prolonging repolarization of phase 3 (slowing down
K+ release).It also suppresses the norepinephrine-induced increase in the sarcoplasmic
reticulum (SR)
Ca2+ leak and the spontaneous SR Ca2+ release,
which are the major triggers for atrial fibrillation.
|
Moxonidine
https://en.wikipedia.org/wiki/Moxonidine

Moxonidine (INN)
is a new-generation alpha-2/imidazoline receptor agonist antihypertensive drug
licensed for the treatment of mild to moderate essential hypertension. It
may have a role when thiazides, beta-blockers, ACE
inhibitors, and calcium
channel blockers are not
appropriate or have failed to control blood pressure. In addition, it demonstrates favourable effects on parameters of the insulin resistance syndrome,
apparently independent of blood pressure reduction. It is also a growth hormone releaser.
Moxonidine is a selective agonist at the imidazoline receptor subtype 1 (I1). This receptor subtype is found in both the rostral ventro-lateral pressor and ventromedial depressor areas of the medulla oblongata. Moxonidine therefore causes a decrease in sympathetic
nervous system activity and, therefore, a decrease in blood pressure.
Compared to the older central-acting antihypertensives, moxonidine binds with much greater affinity to the imidazoline I1-receptor than to the α2-receptor. In contrast, clonidine binds to both receptors with near equal affinity. Moxonidine has an affinity for I1 that is 33 times greater than α2, compared to clonidine which is only four times greater.
In addition, moxonidine may also promote sodium excretion, improve insulin resistance and glucose tolerance and protect against hypertensive target organ damage, such as kidney disease and cardiac hypertrophy.
|
Nadolol
https://en.wikipedia.org/wiki/Nadolol

Nadolol,
sold under the brand name Corgard among
others, is a medication used to treat high blood pressure, heart
pain, and atrial
fibrillation. It
has also been used to prevent migraine headaches and
complications of cirrhosis It
is taken by mouth.
Nadolol is a non-selective beta blocker; that is, it non-selectively blocks both beta-1 and beta-2 receptors. It has a preference for beta-1 receptors, which are predominantly located in the heart, thereby inhibiting the effects of catecholamines and
causing a decrease in heart rate and blood pressure. Its inhibition of beta-2 receptors, which are mainly located in the bronchial smooth muscle of the airways,
leads to airway constriction similar to that seen in asthma. Inhibition of beta-1 receptors in the juxtaglomerular apparatus of the kidney inhibits the renin–angiotensin system, causing a decrease in vasoconstriction and
a decrease in water retention. Nadolol's inhibition of beta-1 receptors in the heart and kidney leads to its effects on lowering blood pressure.
The drug impairs AV node conduction and decreases sinus rate.
Nadolol may also increase plasma triglycerides and decrease HDL-cholesterol levels.
|
Propranolol
https://en.wikipedia.org/wiki/Propranolol

Propranolol,
sold under the brand name Inderal among
others, is a medication of the beta blocker class. It
is used to treat high blood pressure,
a number of types of irregular heart rate, thyrotoxicosis, capillary
hemangiomas, performance
anxiety, and essential
tremors.[1][2][3] It
is used to prevent migraine headaches,
and to prevent further heart problems in those with angina or
previous heart attacks.Propranolol
is a non-selective beta receptor antagonist. It
competes with sympathomimetic neurotransmitters for binding to receptors, which inhibits sympathetic stimulation of the heart. Blockage of neurotransmitter binding to beta 1 receptors on cardiac myocytes inhibits activation of adenylate cyclase, which in turn inhibits cAMP synthesis leading to reduced PKA activation. This results in less calcium influx to cardiac myocytes through voltage gated L-type calcium channels meaning there is a decreased sympathetic effect on cardiac cells, resulting in antihypertensive effects including reduced heart rate and lower arterial blood pressure..
|
S - Metoprolol Succinate
https://en.wikipedia.org/wiki/Metoprolol
Metoprolol,
marketed under the tradename Lopressor among
others, is a selective β1 receptor blocker medication.[3] It
is used to treat high blood pressure, chest
pain due to poor blood flow to the heart, and a number of conditions involving an abnormally
fast heart rate.[3] It
is also used to prevent further heart problems after myocardial infarction and
to prevent headaches in those with migraines.Metoprolol
blocks β1 adrenergic receptors in heart
muscle cells, thereby decreasing the slope of phase 4 in the nodal action potential (reducing Na+ uptake) and prolonging repolarization of phase 3 (slowing down K+ release). It
also suppresses the norepinephrine-induced increase in
the sarcoplasmic reticulum (SR) Ca2+ leak
and the spontaneous SR Ca2+ release, which are the major
triggers for atrial fibrillation. |
| Cardiovascular Energizers |
Coenzyme Q 10
https://en.wikipedia.org/wiki/Coenzyme_Q10

Coenzyme Q, also known as ubiquinone, is a coenzyme family that is ubiquitous in animals and most bacteria (hence the name ubiquinone). In humans, the most common form is Coenzyme Q10 or ubiquinone-10. CoQ10 is not
approved by the U.S. Food and Drug Administration (FDA) for the treatment of any medical condition; however, it is sold as a dietary supplement and is an ingredient in some cosmetics.
It is a 1,4-benzoquinone, where Q refers to the quinone chemical group and 10 refers to the number of isoprenyl chemical subunits in its tail. In natural ubiquinones, the number can be anywhere from 6 to 10. This family of fat-soluble substances, which resemble vitamins,
is present in all respiring eukaryotic cells, primarily in the mitochondria. It is a component of the electron transport chain and participates in aerobic cellular respiration, which generates energy in the form of ATP.
Ninety-five percent of the human body's energy is generated this way.Organs with the highest energy requirements—such as the heart, liver, and kidney—have the highest CoQ10 concentrations.
There are three redox states of CoQ: fully oxidized (ubiquinone), semiquinone (ubisemiquinone), and fully reduced (ubiquinol). The capacity of this molecule to act as a two-electron carrier (moving between the quinone and quinol form) and a one-electron carrier (moving between the semiquinone and one of these other forms) is central to its role in the electron transport chain due to the iron–sulfur
clusters that can only accept one electron at a time, and as a free-radical–scavenging antioxidant.
|
| Cerdiac Glycosides |
Digoxin
https://en.wikipedia.org/wiki/Digoxin
Digoxin,
sold under the brand name Lanoxin among
others, is a medication used to treat various heart conditions. Most
frequently it is used for atrial fibrillation, atrial
flutter, and heart
failure.Digoxin's primary mechanism of action involves inhibition of the sodium potassium adenosine triphosphatase (Na+/K+
ATPase), mainly in the myocardium. This inhibition causes an increase in intracellular sodium levels,
resulting in decreased activity of the sodium-calcium exchanger, which normally imports three extracellular sodium
ions into the cell and transports one intracellular calcium ion out of the cell.
|
| Diuretics |
Frusemide
https://en.wikipedia.org/wiki/Furosemide

Furosemide,
sold under the brand name Lasix among
others, is a medication used to treat fluid build-up due
to heart failure, liver
scarring, or kidney
disease. It
may also be used for the treatment of high blood pressure. It
can be taken by injection into a vein or
by mouth. When taken by
mouth, it typically begins working within an hour, while intravenously, it typically begins working within five minutes..Furosemide, like other loop diuretics, acts by inhibiting the luminal Na-K-Cl
cotransporter in the thick
ascending limb of the loop
of Henle, by binding to the chloride transport channel, thus causing sodium, chloride, and potassium loss in urine.
|
Hydrochlorothiazide
https://en.wikipedia.org/wiki/Hydrochlorothiazide

Hydrochlorothiazide (HCTZ or HCT)
is a diuretic medication
often used to treat high blood pressure and swelling
due to fluid build up. Other
uses include treating diabetes insipidus and renal
tubular acidosis and to
decrease the risk of kidney stones in
those with a high calcium level in the urine. For
high blood pressure it is sometimes considered as a first-line treatment. HCTZ
is taken by mouth and may be combined with other blood pressure medications as
a single pill to increase effectiveness.Hydrochlorothiazide belongs to thiazide class of diuretics.
It reduces blood volume by acting on the kidneys to reduce sodium (Na+)
reabsorption in the distal convoluted tubule. The major site of action in the nephron appears on an electroneutral NaCl co-transporter by competing for the chloride site on the transporter. By impairing Na+ transport
in the distal convoluted tubule, hydrochlorothiazide induces a natriuresis and concomitant water loss. Thiazides increase the reabsorption of calcium in this segment in a manner unrelated to sodium transport. Additionally, by other mechanisms, HCTZ is believed to lower peripheral vascular resistance.
|
Indapamide
https://en.wikipedia.org/wiki/Indapamide

Indapamide is
a thiazide-like diuretic drug generally
used in the treatment of hypertension,
as well as decompensated heart failure.
Combination preparations with perindopril (an ACE
inhibitor antihypertensive)
are also available. Thiazide-like diuretics (indapamide and chlorthalidone) appear to be more effective than the thiazide-type diuretics (hydrochlorothiazide) in reducing risk of major cardiovascular events and heart failure in persons with high blood pressure. In
terms of risk of stroke, both thiazide-type and thiazide-like diuretic are effective in reducing it.Both drug classes also appear to have similar rates of adverse effects when compared to other classes of anti-hypertensives.
|
Telmisartan
https://en.wikipedia.org/wiki/Telmisartan

Telmisartan,
sold under the trade name Micardis among
others, is a medication used to treat high blood pressure, heart
failure, and diabetic
kidney disease. It
is a reasonable initial treatment for high blood press. It
is taken by mouth. Versions
are available as the combination telmisartan/hydrochlorothiazide and
telmisartan/amlodipine..
elmisartan is an angiotensin II receptor blocker that shows high affinity for the angiotensin II receptor type 1 (AT1), with a binding affinity 3000 times greater for AT1 than AT2.
In addition to blocking the RAS, telmisartan acts as a selective modulator of peroxisome proliferator-activated receptor gamma (PPAR-γ), a central regulator of insulin and glucose metabolism. It is believed that telmisartan's dual mode of action may provide protective benefits against the vascular and renal damage caused by diabetes and cardiovascular
disease (CVD).
Telmisartan's activity at the peroxisome proliferator-activated receptor delta (PPAR-δ) receptor has prompted speculation around its potential as a sport doping agent as an alternative to GW 501516. Telmisartan activates PPAR-δ receptors in several tissues.
Also, Telmisartan has a PPARγ agonist activity.
|
Furosemide
https://en.wikipedia.org/wiki/Furosemide
Furosemide, sold under the brand name Lasix among
others, is a medication used to treat fluid build-up due
to heart failure, liver
scarring, or kidney disease.It
may also be used for the treatment of high blood pressure.Furosemide
is primarily used for the treatment of edema,
but also in some cases of hypertension (where there is also kidney or heart impairment).It is often viewed as a first-line agent in most people with edema caused by congestive
heart failure.
Furosemide, like other loop diuretics, acts by inhibiting the luminal Na-K-Cl cotransporter in the thick ascending limb of the loop of Henle, by binding to the chloride transport channel, thus causing sodium, chloride, and potassium loss in urine.
The action on the distal tubules is independent of any inhibitory effect on carbonic anhydrase or aldosterone; it also abolishes the corticomedullary osmotic gradient and blocks negative, as well as positive, free water clearance. Because of the large NaCl absorptive capacity of the loop of Henle, diuresis is not limited by development of acidosis, as it is with the carbonic anhydrase inhibitors.
Additionally, furosemide is a noncompetitive subtype-specific blocker of GABA-A receptors. Furosemide has been reported to reversibly antagonize GABA-evoked currents of α6β2γ2 receptors at μM concentrations, but not α1β2γ2 receptors. During development, the α6β2γ2 receptor increases in expression in
cerebellar granule neurons, corresponding to increased sensitivity to furosemide.
|
| Idiopathic Pulmonary Fibrosis |
Pirfenidone
https://en.wikipedia.org/wiki/Pirfenidone

Pirfenidone is
a medication used for the treatment of idiopathic pulmonary fibrosis.
It works by reducing lung fibrosis through downregulation of the production of growth factors and procollagens I
and II.Pirfenidone has well-established antifibrotic and anti-inflammatory properties
in various in vitro systems and animal models of fibrosis.A number of cell-based studies have shown that pirfenidone reduces fibroblast proliferation,inhibits transforming
growth factor beta stimulated collagen production and reduces the production of fibrogenic mediators such as transforming growth factor beta. Pirfenidone has also been shown to reduce production of inflammatory mediators such as tumor
necrosis factor alpha and IL-1β in both cultured cells and isolated human peripheral
blood mononuclear cells.These activities are consistent with the broader antifibrotic and anti-inflammatory activities observed in animal models of fibrosis..
|
| Peripheral Vasodilators |
Cinnarizine
https://en.wikipedia.org/wiki/Cinnarizine

.Cinnarizine is
an antihistamine and calcium
channel blocker of the diphenylmethylpiperazine group. It
is prescribed for nausea and vomiting due to motion sickness or
other sources such as chemotherapy, vertigo, or Ménière's
disease.Cinnarizine is classified as a selective antagonist of T-type voltage-operated calcium ion channels, because its binding blocks the channels and keeps them inert. It has a Ki (inhibitory constant) value of 22nM.[ It is also known to have antihistaminic, antiserotoninergic and antidopaminergic effects, binding
to H1 histamine receptors, and dopaminergic (D2) receptors.
|
Ginkgo Biloba
https://en.wikipedia.org/wiki/Ginkgo_biloba
Ginkgo biloba,
conly known as ginkgo or gingko (both
pronounced ),
also known as the maidenhair tree, is
the only living species in the division Ginkgophyta,
all others being extinct. It is a very old species, with some fossils dating back 270 million years. Native to China, the
tree is widely cultivated, and was cultivated early in human history.
It has various uses in traditional medicine and
as a source of food.
|
Papaverine HCl
https://en.wikipedia.org/wiki/Papaverine

Papaverine (Latin papaver,
"poppy") is an opium alkaloid antispasmodic drug,
used primarily in the treatment of visceral spasm and vasospasm (especially
those involving the intestines, heart, or brain), and occasionally in the treatment of erectile dysfunction.
It is used in the treatment of acute mesenteric ischemia.
While it is found in the opium poppy,
papaverine differs in both structure and pharmacological action from the analgesic morphine-like compounds.
The in vivo mechanism of action is not entirely clear, but an inhibition of the enzyme phosphodiesterase causing elevation of cyclic AMP and cyclic GMP[clarification needed] levels
is significant. It may also alter mitochondrial respiration.
Papaverine has also been demonstrated to be a selective phosphodiesterase inhibitor for the PDE10A subtype found mainly in the striatum of the brain. When administered chronically to mice, it produced motor and cognitive deficits and increased anxiety, but conversely may produce an antipsychotic effect, although
not all studies support this view.
|
| Vasopressors |
Phenylephrine
https://en.wikipedia.org/wiki/Phenylephrine

Phenylephrine is
a medication primarily used as a decongestant,
to dilate the pupil,
to increase blood pressure,
and to relieve hemorrhoids. While
marketed as a decongestant, taken by mouth at recommended doses it is of unclear benefit for hay fever..
Phenylephrine is a sympathomimetic drug, which means that it mimics the actions of epinephrine (commonly known as adrenaline) or norepinephrine. Phenylephrine selectively binds to alpha-1 receptors which cause blood vessels to constrict.
Whereas pseudoephedrine causes both vasoconstriction and increase of mucociliary clearance through its nonspecific adrenergic activity, phenylephrine's selective α-adrenergic agonism causes vasoconstriction alone, creating a difference in their methods of action.
|
| |
|
| Central Nervous System |
| Anti-Emetics & Antinauseants |
Domperidone
https://en.wikipedia.org/wiki/Domperidone

Domperidone, sold under the brand name Motilium among others, is a medication used as an antiemetic, gastric prokinetic agent, and galactagogue. It may be taken by mouth or rectally,
and is available as a tablet, orally disintegrating tablets, suspension, and suppositories.[The
drug is used to relieve nausea and vomiting; to increase the transit of food through the stomach (by increasing gastrointestinal peristalsis); and to promote lactation (breast
milk production) by release of prolactin.
It is a peripherally selective dopamine D2 receptor antagonist
Domperidone is a peripherally
selective dopamine D2 and D3 receptor antagonist. It
has no clinically significant interaction with the D1 receptor,
unlike metoclopramideThe
medication provides relief from nausea by blocking D receptors. It
blocks dopamine receptors in the anterior pituitary gland
increasing release of prolactin which
in turn increases lactation. Domperidone
may be more useful in some patients and cause harm in others by way of the genetics of
the person, such as polymorphisms in
the drug transporter gene ABCB1 (which
encodes P-glycoprotein),
the voltage-gated potassium channel KCNH2 gene
(hERG/Kv11.1),
and the α1D—adrenoceptor ADRA1D gene.
|
Fosaprepitant
https://en.wikipedia.org/wiki/Fosaprepitant

Fosaprepitant (Emend for Injection (US), Ivemend (EU)) is an antiemetic drug, administered intravenously. It is a prodrug of aprepitant.
Fosaprepitant was developed by Merck & Co. and was approved by the United States Food and Drug Administration (FDA) on January 25, 2008 and by the European Medicines Agency (EMA) on January
11 of the same year.
|
Granisetron
https://en.wikipedia.org/wiki/Granisetron

Granisetron is
a serotonin 5-HT3 receptor
antagonist used as an antiemetic to
treat nausea and
vomiting following chemotherapy and
radiotherapy. Its main effect is to reduce the activity of the vagus nerve,
which is a nerve that activates the vomiting center in the medulla oblongata.
It does not have much effect on vomiting due to motion sickness. This drug does not have any effect on dopamine receptors
or muscarinic receptors..
|
Metoclopramide
https://en.wikipedia.org/wiki/Metoclopramide

Metoclopramide is
a medication used mostly for stomach and esophageal problems. It
is commonly used to treat and prevent nausea and vomiting,
to help with emptying of the stomach in
people with delayed stomach emptying,
and to help with gastroesophageal reflux disease. It
is also used to treat migraine headaches.
The antiemetic action of metoclopramide is due to its antagonist activity at D2 receptors in the chemoreceptor trigger zone in the central nervous system — this action prevents nausea and vomiting triggered by most stimuli. At higher doses, 5-HT3 antagonist activity may also contribute to the antiemetic effect.
The gastroprokinetic activity of metoclopramide is mediated by muscarinic activity, D2 receptor antagonist activity, and 5-HT4 receptor agonist activity. The gastroprokinetic effect itself may also contribute to the antiemetic effect. Metoclopramide also increases the tone of the lower esophageal sphincter.
Metoclopramide might influence on mood because of its blockade action on 5-HT4 and 5-HT3.
|
Ondansetron Di HCl
https://en.wikipedia.org/wiki/Ondansetron

Ondansetron,
sold under the brand name Zofran, is a medication used to
prevent nausea and vomiting caused
by cancer chemotherapy, radiation
therapy, or surgery. It
is also effective for treating gastroenteritis. It
is ineffective for treating vomiting caused by motion
sickness.Ondansetron is a highly
specific and selective serotonin 5-HT3 receptor
antagonist, with low
affinity for dopamine receptors. The 5-HT3 receptors are present both peripherally on vagal
nerve terminals and centrally in the chemoreceptor trigger zone of the area
postrema in the medulla. Serotonin is released by the enterochromaffin cells of the small intestine in response to chemotherapeutic agents and may stimulate vagal afferents (via 5-HT3 receptors) to initiate the vomiting reflex. It is thought that ondansetron's antiemetic action is mediated mostly via antagonism of vagal afferents with a minor contribution from antagonism of central receptors.
|
Prochlorperazine
https://en.wikipedia.org/wiki/Prochlorperazine
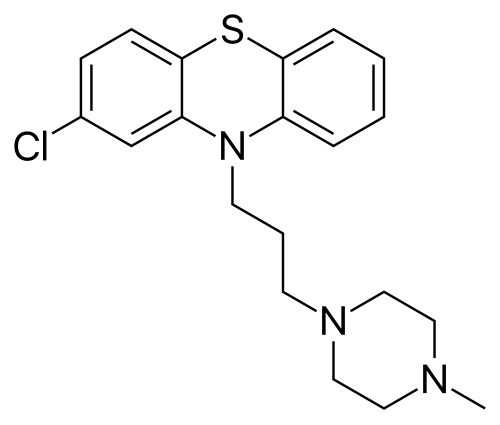
Prochlorperazine,
sold under the brand name Compazine among others, is
Prochlorperazine is thought to exert its antipsychotic effects by blocking dopamine receptors.
Prochlorperazine is analogous to chlorpromazine; both of these agents antagonize dopaminergic D2 receptors in various pathways of the central nervous system. This D2 blockade
results in antipsychotic, antiemetic and other effects. Hyperprolactinemia is a side effect of dopamine antagonists as blockade of D2 receptors within the tuberoinfundibular
pathway results in increased plasma levels of prolactin due to increased secretion by lactotrophs in the anterior pituitary.
a medication used to treat nausea, schizophrenia, migraines,
and anxiety.It is a less preferred
medication for anxiety. It may be taken by mouth, rectally, injection
into a vein, or injection
into a muscle..
|
Trifluoperazine
https://en.wikipedia.org/wiki/Trifluoperazine
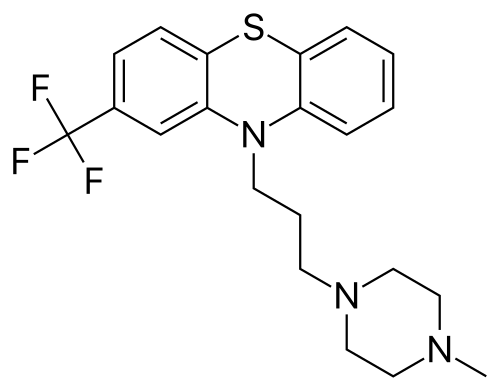
Trifluoperazine,
sold under a number of brand names, is a typical antipsychotic primarily
used to treat schizophrenia. It
may also be used short term in those with generalized anxiety disorder but
is less preferred to benzodiazepines. It
is of the phenothiazine chemical
class.Trifluoperazine has central antiadrenergic,[antidopaminergic, and
minimal anticholinergic effects. It is believed to work by
blockading dopamine D1 and D2 receptors
in the mesocortical and mesolimbic
pathways, relieving or
minimizing such symptoms of schizophrenia as hallucinations, delusions,
and disorganized thought and speech.
|
Trimethobenzamide
https://en.wikipedia.org/wiki/Trimethobenzamide

Trimethobenzamide (trade
names Tebamide, Tigan)
is an antiemetic used
to prevent nausea and vomiting.Trimethobenzamide
is an antagonist of
the D2 receptor. It
is believed to affect the chemoreceptor trigger zone (CTZ) of the medulla
oblongata to suppress nausea and vomiting.
|
| Antiarrhythmic Agent |
Haloperidol
https://en.wikipedia.org/wiki/Haloperidol

Haloperidol,
sold under the brand name Haldol among
others, is a typical antipsychotic medication.[4] Haloperidol
is used in the treatment of schizophrenia,
tics in Tourette syndrome, mania in bipolar
disorder, nausea and vomiting, delirium,
agitation, acute psychosis,
and hallucinations in alcohol
withdrawal.Haloperidol is heavily protein bound in human plasma, with a free fraction of only 7.5 to 11.6%. It is also extensively metabolized in the liver with only about 1% of the administered dose excreted unchanged in the urine. The greatest proportion of the hepatic clearance is by glucuronidation, followed by reduction and CYP-mediated oxidation, primarily by CYP3A4.
|
| Anticovulsants |
Carbamazepine
https://en.wikipedia.org/wiki/Carbamazepine

Carbamazepine (CBZ),
sold under the trade name Tegretol among
others, is an anticonvulsant medication used
primarily in the treatment of epilepsy and neuropathic
pain. It
is used in schizophrenia along
with other medications and as a second-line agent in bipolar disorder. Carbamazepine
appears to work as well as phenytoin and valproate for
focal and generalized seizures. It
is not effective for absence or myoclonic
seizures..Carbamazepine
is a sodium channel blocker. It
binds preferentially to voltage-gated sodium channels in their inactive conformation, which prevents repetitive and sustained firing of an action potential. Carbamazepine has effects on serotonin systems but the relevance to its antiseizure effects is uncertain. There is evidence that it is a serotonin releasing agent and
possibly even a serotonin reuptake inhibitor.
|
Divalproex
https://en.wikipedia.org/wiki/Valproate

Valproate (VPA)
and its valproic acid, sodium
valproate, and valproate semisodium forms
are medications primarily used to treat epilepsy and bipolar
disorder and prevent migraine
headaches. They
are useful for the prevention of seizures in those with absence seizures, partial
seizures, and generalized
seizures.Although the mechanism of action of valproate is not fully understood,[50] traditionally,
its anticonvulsant effect has been attributed to the blockade of voltage-gated sodium channels and increased brain levels of gamma-aminobutyric
acid (GABA). The GABAergic effect is also believed to contribute towards the anti-manic properties of valproate.[50] In animals, sodium valproate raises cerebral and cerebellar levels of the inhibitory synaptic neurotransmitter, GABA, possibly by inhibiting GABA degradative enzymes,
such as GABA transaminase, succinate-semialdehyde
dehydrogenase and by inhibiting the re-uptake of GABA by neuronal cells.
|
Felbamate
https://en.wikipedia.org/wiki/Felbamate

Felbamate (marketed
under the brand name Felbatol by MedPointe)
is an anticonvulsant used
in the treatment of epilepsy.
It is used to treat partial seizures (with
and without generalization) in adults and partial and generalized seizures associated with Lennox–Gastaut syndrome in
children. However, an increased risk of potentially fatal aplastic anemia and/or liver
failure limit the drug's
usage to severe refractory epilepsy.Felbamate has been proposed to a unique dual mechanism of action as a positive modulator of GABAA receptors and
as a blocker of NMDA receptors, particularly isoforms containing the NR2B subunit. Although
it is clear that felbamate does cause pharmacological inhibition of NMDA receptors, the relevance of NMDA receptor blockade as a strategy for the treatment of human epilepsy has been questioned. Therefore, the importance of the effects of felbamate on NMDA receptors to its therapeutic action in epilepsy is uncertain.
|
Fosphenytoin Sodium
https://en.wikipedia.org/wiki/Fosphenytoin
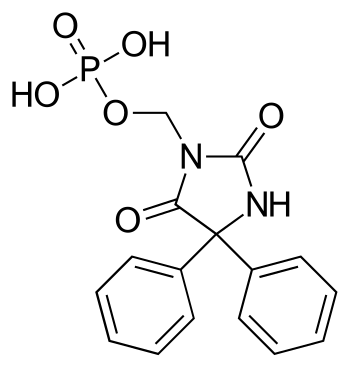
Fosphenytoin (fosphenytoin sodium,
trade names Cerebyx, Parke-Davis; Prodilantin, Pfizer
Holding France) is a water-soluble phenytoin prodrug that
is administered intravenously to deliver phenytoin, potentially more safely than intravenous phenytoin.
It is most commonly used in the acute treatment of convulsive status epilepticus.One millimole of
phenytoin is produced for every millimole of fosphenytoin administered; the hydrolysis of fosphenytoin also yields phosphate and formaldehyde,
the latter of which is subsequently metabolized to formate, which is in turn metabolized by a folate dependent mechanism.
|
Gabapentin
https://en.wikipedia.org/wiki/Gabapentin

Gabapentin,
sold under the brand name Neurontin among
others, is an anticonvulsant medication
used to treat partial seizures, neuropathic
pain, hot
flashes, and restless
legs syndrome. It
is recommended as one of a number of first-line medications for the treatment of neuropathic pain caused by diabetic neuropathy, postherpetic
neuralgia, and central neuropathic pain.About 15% of those given gabapentin for diabetic neuropathy or postherpetic neuralgia have a measurable benefit. Gabapentin
is taken by mouth.Gabapentin
is a gabapentinoid, or a ligand of
the auxiliary α2δ
subunit site
of
certain voltage-dependent calcium channels (VDCCs), and thereby acts as an inhibitor of
α2δ subunit-containing VDCCs.There are two drug-binding α2δ subunits, α2δ-1 and α2δ-2,
and gabapentin shows similar affinity for (and hence lack of selectivity between)
these two sites. Gabapentin is selective in its binding to the α2δ VDCC subunit. Despite the fact that gabapentin is a GABA
analogue, and in spite of its name, it does not bind to the GABA receptors, does not convert into GABA or
another GABA receptor agonist in
vivo, and does not modulate GABA transport or metabolism.There
is currently no evidence that the effects of gabapentin are mediated by any mechanism other than inhibition of α2δ-containing
VDCCs.In accordance, inhibition of α2δ-1-containing VDCCs by gabapentin appears to be responsible for its anticonvulsant, analgesic,
and anxiolytic effects.
|
Lamotrigine
https://en.wikipedia.org/wiki/Lamotrigine

Lamotrigine, sold as the brand name Lamictal among
others, is an anticonvulsant medication used
to treat epilepsy and
to delay or prevent the recurrence of depressive episodes in bipolar disorder. For
epilepsy, this includes focal seizures, tonic-clonic
seizures, and seizures in Lennox-Gastaut
syndrome. In
bipolar disorder, lamotrigine has not been shown to reliably treat acute depression; but for patients with bipolar disorder who are not currently symptomatic, it appears to be effective in reducing the risk of future episodes of depression.It is a triazine derivate that inhibits voltage-sensitive sodium
channels, leading to stabilization of neuronal membranes. It also blocks L-, N-, and P-type calcium channels and weakly inhibits the serotonin 5-HT3 receptor. These
actions are thought to inhibit release of glutamate at cortical projections in the ventral
striatum limbic areas, and its neuroprotective and
antiglutamatergic effects have been pointed out as promising contributors to its mood stabilizing activity. Observations that lamotrigine reduced γ-aminobutyric acid (GABA) A receptor-mediated neurotransmission in rat amygdala, suggest that a GABAergic mechanism may also be involved. It appears that lamotrigine does not increase GABA blood levels in humans.
|
Levetiracetam
https://en.wikipedia.org/wiki/Levetiracetam

Levetiracetam,
sold under the brand name Keppra among
others, is a medication used to treat epilepsy. It
is used for partial-onset, myoclonic,
or tonic–clonic seizures
and is taken either by mouth as
an immediate or extended
release formulation or by injection
into a vein.The exact mechanism by which levetiracetam acts to treat epilepsy is unknown. Levetiracetam does not exhibit pharmacologic actions similar to that of classical anticonvulsants. It does not inhibit voltage-dependent Na+ channels, does not affect GABAergic transmission, and does not bind to GABAergic or glutamatergic receptors. However,
the drug binds to SV2A, a synaptic
vesicle glycoprotein, and inhibits presynaptic calcium
channels, reducing neurotransmitter
release and acting as a neuromodulator. This is believed to impede impulse conduction across synapses.
|
Phenytoin
https://en.wikipedia.org/wiki/Phenytoin

Phenytoin (PHT),
sold under the brand name Dilantin among
others, is an anti-seizure
medication. It
is useful for the prevention of tonic-clonic seizures and focal
seizures, but not absence
seizures. The
intravenous form is used for statusPhenytoin
is believed to protect against seizures by causing voltage-dependent block of voltage
gated sodium channels. This
blocks sustained high frequency repetitive firing of action
potentials. This is accomplished by reducing the amplitude of sodium-dependent action potentials through enhancing steady state inactivation. Sodium channels exist in three main conformations: the resting state, the open state, and the inactive state. epilepticus that
does not improve with benzodiazepines. It
may also be used for certain heart arrhythmias or neuropathic
pain.
|
Pregabalin
https://en.wikipedia.org/wiki/Pregabalin

Pregabalin, marketed under the brand name Lyrica among
others, is a medication used to treat epilepsy, neuropathic
pain, fibromyalgia, restless
leg syndrome, and generalized anxiety disorder. Its
use in epilepsy is as an add-on therapy for partial seizures.Pregabalin
is a gabapentinoid and
acts by inhibiting certain calcium
channels. Specifically
it is a ligand of
the auxiliary α2δ
subunit site of certain voltage-dependent
calcium channels (VDCCs), and thereby acts as an inhibitor of
α2δ
subunit-containing VDCCs. |
Retigabine
https://en.wikipedia.org/wiki/Retigabine

Retigabine (INN)
or ezogabine (USAN)
is an anticonvulsant used
as an adjunctive treatment for partial epilepsies in
treatment-experienced adult patients.Retigabine acts as a neuronal KCNQ/Kv7 potassium
channel opener, a mechanism of action markedly different from that of any current anticonvulsants. This mechanism of action is similar to that of the chemically-similar flupirtine, which
is used mainly for its analgesic properties.
|
Sodium Valproate
https://en.wikipedia.org/wiki/Valproate
Valproate (VPA)
and its valproic acid, sodium
valproate, and valproate semisodium forms
are medications primarily used to treat epilepsy and bipolar
disorder and prevent migraine
headaches..Valproate's
precise mechanism of action is unclear. Proposed
mechanisms include affecting GABA levels,
blocking voltage-gated sodium channels,
and inhibiting histone deacetylases. Valproic
acid is a branched short-chain fatty acid (SCFA)
made from valeric acid.
|
Topiramate
https://en.wikipedia.org/wiki/Topiramate

Topiramate,
sold under the brand name Topamax among
others, is a medication used to treat epilepsy and
prevent migraines. It
has also been used in alcohol dependence. For
epilepsy this includes treatment for generalized or focalSeveral
cellular targets have been proposed to be relevant to the therapeutic activity of topiramate. These include (1) voltage-gated sodium
channels;
(2) high-voltage-activated calcium
channels;
(3) GABA-A
receptors;
(4) AMPA/kainate receptors; and (5) carbonic
anhydrase isoenzymes.
There is evidence that topiramate may alter the activity of its targets by modifying their phosphorylation state instead of by a direct action.[42] The
effect on sodium channels could be of particular relevance for seizure protection. Although topiramate does inhibit high-voltage-activated calcium channels, the relevance to clinical activity is uncertain. Effects on specific GABA-A receptor isoforms could also contribute to the antiseizure activity of the drug. Topiramate selectively inhibits cytosolic (type II) and membrane associated (type IV) forms of carbonic anhydrase. The action on carbonic anhydrase isoenzymes may contribute to the drug's side-effects, including its propensity to cause metabolic acidosis and calcium phosphate kidney stones. seizures. It
is taken by mouth.
|
Valproic Acid
https://en.wikipedia.org/wiki/Valproate
Valproate (VPA)
and its valproic acid, sodium
valproate, and valproate semisodium forms
are medications primarily used to treat epilepsy and bipolar
disorder and prevent migraine
headaches..Valproate's
precise mechanism of action is unclear. Proposed
mechanisms include affecting GABA levels,
blocking voltage-gated sodium channels,
and inhibiting histone deacetylases. Valproic
acid is a branched short-chain fatty acid (SCFA)
made from valeric acid.
|
Vigabatrin
https://en.wikipedia.org/wiki/Vigabatrin
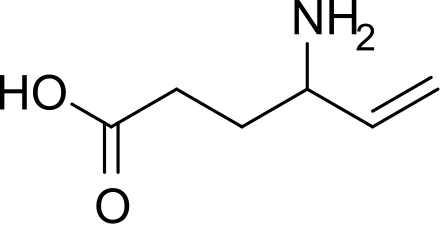
Vigabatrin, brand name Sabril, is a medication used to treat epilepsy. It became available as a generic medication in 2019.
It works by inhibiting the breakdown of γ-aminobutyric acid (GABA). It is also known as γ-vinyl-GABA, and is a structural analogue of GABA, but does not bind to GABA receptors.Vigabatrin
is an irreversible mechanism-based inhibitor of gamma-aminobutyric
acid aminotransferase (GABA-AT),
the enzyme responsible
for the catabolism of GABA.
Inhibition of GABA-AT results in increased levels of GABA in
the brain. Vigabatrin
is a racemic compound,
and its [S]-enantiomer is
pharmacologically active.
|
| Antimanic Drugs |
Haloperidol
https://en.wikipedia.org/wiki/Haloperidol
Haloperidol is a typical butyrophenone type
antipsychotic that exhibits high affinity dopamine D2 receptor
antagonism and slow receptor dissociation kinetics. It has effects similar
to the phenothiazines.Haloperidol,
sold under the brand name Haldol among others, is a typical
antipsychotic medication. Haloperidol is used in the treatment of schizophrenia,
tics in Tourette syndrome, mania in bipolar
disorder, nausea and vomiting, delirium, agitation, acute psychosis,
and hallucinations in alcohol
withdrawal.
|
| |
Quetiapine Fumarate
https://en.wikipedia.org/wiki/Quetiapine

Quetiapine,
sold under the brand name Seroquel among
others, is an atypical antipsychotic medication
used for the treatment of schizophrenia, bipolar
disorder, and major
depressive disorder. Despite
being widely used as a sleep aid due its sedating effect,
the benefits of such use do not appear to generally outweigh the side effects.
Quetiapine has the following pharmacological actions:
5-HT1A receptor partial agonist, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT6,
and 5-HT7 receptor antagonist, and 5-HT1B, 5-HT1D, 5-HT1E,
and 5-HT1F receptor ligand
α1- and α2-adrenergic receptor antagonist
Histamine H1 receptor antagonist
Muscarinic acetylcholine receptor antagonist
|
| Antipsychotics |
Aripiprazole
https://en.wikipedia.org/wiki/Aripiprazole

Aripiprazole, sold under
the brand name Abilify among
others, is an atypical antipsychotic. It
is primarily used in the treatment of schizophrenia and bipolar
disorder. Other
uses include as an add-on treatment in major
depressive disorder, tic
disorders and irritability
associated with autism..
Aripiprazole's mechanism
of action is different from
those of the other FDA-approved atypical antipsychotics (e.g., clozapine, olanzapine, quetiapine, ziprasidone,
and risperidone). It
shows differential engagement at
the dopamine receptor (D2).
It appears to show
predominantly antagonist activity on postsynaptic D2 receptors
and partial agonist activity on
presynaptic D2 receptors,
D3 and partially D4)
and is a partial activator of serotonin (5-
HT1A,
5-HT2A, 5-HT2B, 5-HT6,
and 5-HT7).
|
Iloperidone
https://en.wikipedia.org/wiki/Iloperidone

Iloperidone,
commonly known as Fanapt and
previously known as Zomaril, is an atypical
antipsychotic for the
treatment of schizophrenia.Iloperidone
is a monoamine directed towards acting upon and antagonizing specific neurotransmitters, particularly multiple dopamine and serotonin receptor
subtypes. It is considered an ‘atypical’ antipsychotic because it displays serotonin receptor antagonism, similar to other atypical antipsychotics. The older typical antipsychotics are primarily dopamine antagonists.
|
Lurasidone
https://en.wikipedia.org/wiki/Lurasidone

Lurasidone, sold under the trade name Latuda among others, is an antipsychotic medication
used to treat schizophrenia and bipolar
disorder In bipolar it may be used together with a mood stabilizer such
as lithium or valproate. It
is taken by mouth.Lurasidone acts as an antagonist of the dopamine D2 and D3 receptors, the serotonin 5-HT2A and 5-HT7 receptors, and the α2C-adrenergic receptor, and as a partial agonist of the serotonin 5-HT1A receptor. It has only low and likely clinically unimportant affinity for
the serotonin 5-HT2C receptor, which may underlie its low propensity for appetite stimulation and weight
gain. The drug also has negligible affinity for the histamine H1 receptor and the muscarinic acetylcholine receptors, and hence has no antihistamine or anticholinergic effects.
|
Zotepine
https://en.wikipedia.org/wiki/Zotepine

Zotepine is an atypical antipsychotic drug indicated for acute and chronic schizophrenia.
The antipsychotic effect of zotepine is thought to be mediated through antagonist activity
at dopamine and serotonin
receptors. Zotepine has a high affinity for
the D1 and D2 receptors.
It also affects the 5-HT2A, 5-HT2C, 5-HT6,
and 5-HT7 receptors. In
addition, its active metabolite, norzotepine, serves as a potent norepinephrine reuptake inhibitor..
|
| Drugs to control Rigidity & Tremors |
Selegiline
https://en.wikipedia.org/wiki/Selegiline
Selegiline, also known as L-deprenyl and sold under the brand names Eldepryl and Emsam among others, is a medication which is used in the treatment of Parkinson's disease and major depressive disorder.It is provided in the form of a capsule or tablet taken by
mouth for Parkinson's disease and as a patch applied to skin for depression.
Selegiline acts as a monoamine oxidase inhibitor, and increases levels of monoamine neurotransmitters in the brain. At typical clinical doses, selegiline is a selective and irreversible
inhibitor of monoamine oxidase B (MAO-B), increasing levels of dopamine in the brain. In larger doses, it loses its specificity and also inhibits MAO-A, increasing levels of serotonin, norepinephrine,
and dopamine in the brain.
|
| Drugs used to treat Migraine |
Flunarizine
https://en.wikipedia.org/wiki/Flunarizine

Flunarizine,
sold under the brand name Sibelium among
others, is a drug classified as a calcium antagonist which
is used for various indications.Flunarizine is a selective calcium antagonist with moderate other actions including antihistamine, serotonin
receptor blocking and dopamine
D2 blocking
activity. Compared to other calcium channel blockers such as dihydropyridine derivatives, verapamil and diltiazem,
flunarizine has low affinity to voltage-dependent calcium channels. It has been theorised that it may act not by inhibiting calcium entry into cells, but rather by an intracellular mechanism
such as antagonising calmodulin, a calcium binding protein. |
Frovatriptan
https://en.wikipedia.org/wiki/Frovatriptan

Frovatriptan (trade
name Frova) is a triptan drug developed
by Vernalis for
the treatment of migraine headaches and
for short term prevention of menstrual miraine.Frovatriptan
is a 5HT receptor agonist,
with high affinity for the 5-HT1B/1D receptors. It has no significant effects on the GABAA mediated
channel activity and benzodiazepine binding sites.
|
Sumatriptan
https://en.wikipedia.org/wiki/Sumatriptan

Sumatriptan,
sold under the brand name Imitrex among
others, is a medication used to treat migraine headaches and cluster
headaches.
Sumatriptan is structurally similar to serotonin (5-HT), and is a 5-HT receptor (types 5-HT1D and 5-HT1B) agonist.
Sumatriptan's primary therapeutic effect, however, is in its inhibition of the release of Calcitonin gene-related peptide (CGRP), likely through its 5-HT1D/1B receptor-agonist action. This is substantiated by the efficacy of newly developed CGRP antagonists and antibodies in the preventative treatment of migraine. However, how agonism of the 5-HT1D/1B receptors inhibits CGRP release is not fully understood. CGRP is believed to cause sensitization of trigeminal nociceptive neurons, contributing to the pain experienced in migraine.
Sumatriptan is also shown to decrease the activity of the trigeminal nerve, which presumably accounts for sumatriptan's efficacy in treating cluster headaches. The injectable form of the drug has been shown to abort a cluster headache within 30 minutes in 77% of cases.
|
| Drugs used in Peripheral Neuropathy |
Benfotiamine
https://en.wikipedia.org/wiki/Benfotiamine

Benfotiamine (rINN,
or S-benzoylthiamine O-monophosphate)
is a synthetic S-acyl derivative of thiamine (vitamin
B1) that sold as a medication or dietary supplement to
treat diabetic neuropathy. Combination
drugs with pyridoxine or cyanocobalamin are
also marketed.
Benfotiamine is more bioavailable than thiamine salts, providing higher levels of thiamine in muscle, brain, liver, and kidney.
Benfotiamine is dephosphorylated to S-benzoylthiamine by ecto-alkaline phosphatases present in the intestinal mucosa, and is then hydrolyzed to thiamine by thioesterases in the liver.
Benfotiamine mainly acts on peripheral tissues through an increase in transketolase activity.
|
Mecobalamin / Methylcobalamin
https://en.wikipedia.org/wiki/Methylcobalamin

Methylcobalamin (mecobalamin, MeCbl, or MeB12) is a cobalamin, a form of vitamin B12. It differs from cyanocobalamin in that the cyano
group at the cobalt is replaced with a methyl group. Methylcobalamin features an octahedral cobalt(III) centre and can be obtained as bright red crystals. From the perspective of coordination chemistry, methylcobalamin is notable as a rare example of a compound that contains metal–alkyl bonds. Nickel–methyl intermediates have been proposed for the final step of methanogenesis.
Methylcobalamin is equivalent physiologically to vitamin B12,and can be used to prevent or treat pathology arising from a lack of vitamin B12 intake (vitamin B12 deficiency).
Methylcobalamin is also used in the treatment of peripheral neuropathy, diabetic neuropathy, and as a preliminary treatment for amyotrophic lateral sclerosis.
Methylcobalamin that is ingested is not used directly as a cofactor, but is first converted by MMACHC into cob(II)alamin. Cob(II)alamin is then later converted into the other 2 forms, adenosylcobalamin and methylcobalamin for use as cofactors. That is, methylcobalamin is first dealkylated and then regenerated.
According to one author, it is important to treat vitamin B12 deficiency with hydroxocobalamin or cyanocobalamin or a combination of adenosylcobalamin and
methylcobalamin, and not methylcobalamin alone.
|
| Hepatic Encephalopathy |
Amino Acids
https://en.wikipedia.org/wiki/Amino_acid

Amino acids are organic
compounds that contain amine (–NH2)
and carboxyl (–COOH) functional
groups, along with a side
chain (R group) specific to each amino acid. The
key elements of
an amino acid are carbon (C), hydrogen (H), oxygen (O),
and nitrogen (N),
although other elements are found in the side chains of certain amino acids. About 500 naturally occurring amino acids are known (though only 20 appear in the genetic code)
and can be classified in many ways.When taken up into the human body from the diet, the 20 standard amino acids either are used to synthesize proteins, other biomolecules, or are oxidized to urea and carbon
dioxide as a source of energy. The
oxidation pathway starts with the removal of the amino group by a transaminase; the amino group is then fed into the urea
cycle. The other product of transamidation is a keto acid that
enters the citric acid cycle. Glucogenic
amino acids can also be converted into glucose, through gluconeogenesis..
|
| Insomnia |
Eszopiclone
https://en.wikipedia.org/wiki/Eszopiclone

Eszopiclone, sold under the brand-name Lunesta among
others, is a medication used in the treatment of insomnia. Evidence
supports slight to moderate benefit up to six months. It is taken by mouth.Eszopiclone acts on benzodiazepine binding
site situated on GABAA neurons as
a positive allosteric modulator.
|
Ramelteon
https://en.wikipedia.org/wiki/Ramelteon

Ramelteon, sold under the brand name Rozerem among
others, is a sleep agent medication that selectively binds to the MT1 and MT2 receptors in
the suprachiasmatic nucleus (SCN),
instead of binding to GABAA receptors,
such as with drugs like zolpidem.
Ramelteon is a melatonin receptor agonist with both high affinity for melatonin MT1 and MT2 receptors and selectivity over
the MT3 receptor. Ramelteon demonstrates full agonist activity in vitro in cells expressing human MT1 or MT2 receptors, and high selectivity for human MT1 and MT2 receptors compared to the MT3 receptor.
The activity of ramelteon at the MT1 and MT2 receptors is believed to contribute to its sleep-promoting properties, as these receptors, acted upon by endogenous melatonin, are thought to be involved in the maintenance of the circadian rhythm underlying the normal sleep-wake cycle. Ramelteon has no appreciable affinity for the GABA receptor complex or for receptors that bind neuropeptides, cytokines, serotonin, dopamine, noradrenaline, acetylcholine,
and opioids. Ramelteon also does not interfere with the activity of a number of selected enzymes in a standard panel.
|
Zolpidem Tartrate
https://en.wikipedia.org/wiki/Zolpidem

Zolpidem, sold under the brand name Ambien, among
others, is a medication primarily used for the short-term treatment of sleeping
problems.Zolpidem is a ligand of high-affinity positive modulator sites
of GABAA receptors,
which enhances GABAergic inhibition of neurotransmission in the central nervous system. It selectively binds to α1 subunits of
this pentameric ion
channel. Accordingly, it has strong hypnotic properties and weak anxiolytic, myorelaxant,
and anticonvulsant properties. Opposed to diazepam,
zolpidem is able to bind to binary αβ GABA receptors, where it was shown to bind to the α1–α1 subunit interface.
|
| Multiple Sclerosis |
Fingolimod
https://en.wikipedia.org/wiki/Fingolimod

Fingolimod (INN,
trade name Gilenya, Novartis) is an immunomodulating drug,
mostly used for treating multiple sclerosis (MS).Fingolimod is a sphingosine-1-phosphate
receptor modulator, which sequesters lymphocytes in lymph nodes, preventing them from contributing to an autoimmune reaction. It has been reported to reduce the rate of relapses in relapsing-remitting multiple sclerosis by approximately one-half over a two-year period.The molecular biology of phospho-fingolimod is thought to lie in its activity at one of the five sphingosine-1-phosphate receptors, S1PR1. Phospho-fingolimod causes the internalization of
S1P receptors, which sequesters lymphocytes in lymph nodes, preventing them from moving to the central nervous system and causing a relapse of multiple
sclerosis.
|
| Neuroleptic Agent |
Amisulpride
https://en.wikipedia.org/wiki/Amisulpride
Amisulpride is an antiemetic and
antipsychotic medication used at lower doses intravenously to prevent and treat postoperative nausea and vomiting; and at higher doses orally and intramuscularly to
treat schizophrenia and acute psychotic episodes.Amisulpride
functions primarily as a dopamine D2 and D3 receptor antagonist.
It has high affinity for these
receptors with dissociation constants of
3.0 and 3.5 nM, respectively. Although standard doses used to treat psychosis inhibit dopaminergic neurotransmission,
low doses preferentially block inhibitory presynaptic autoreceptors.
|
| Neuropathic Pain |
Carbamazepine
https://en.wikipedia.org/wiki/Carbamazepine
Carbamazepine (CBZ),
sold under the trade name Tegretol among others, is an anticonvulsant
medication used primarily in the treatment of epilepsy and neuropathic
pain. It is used in schizophrenia along
with other medications and as a second-line agent in bipolar disorder.Carbamazepine is a sodium
channel blocker. It binds preferentially to voltage-gated sodium channels in their inactive conformation, which prevents repetitive and sustained firing of an action potential. Carbamazepine has effects on serotonin systems but the relevance to its antiseizure effects is uncertain. There is evidence that it is a serotonin
releasing agent and possibly even a serotonin reuptake inhibitor..
|
| Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) |
Ibuprofen
https://en.wikipedia.org/wiki/Ibuprofen

Ibuprofen is a medication in the nonsteroidal
anti-inflammatory drug (NSAID) class that is used for treating pain, fever,
and inflammation. This includes painful
menstrual periods, migraines,
NSAIDs such as ibuprofen work by inhibiting the cyclooxygenase (COX) enzymes, which convert arachidonic acid to prostaglandin
H2 (PGH2). PGH2, in turn, is converted by other enzymes to several other prostaglandins (which are mediators of pain, inflammation, and fever) and to thromboxane A2 (which stimulates platelet aggregation,
leading to the formation of blood clots).
Like aspirin and indomethacin, ibuprofen is a nonselective COX inhibitor, in that it inhibits two isoforms of cyclooxygenase, COX-1 and COX-2.
and rheumatoid arthritis. It
may also be used to close a patent ductus arteriosus in a premature
baby.It can be used by mouth or intravenously.
|
Nabumetone
https://en.wikipedia.org/wiki/Nabumetone

Nabumetone is a nonacidic NSAID that is rapidly metabolized
in the liver to a major active metabolite, 6-methoxy-2-naphthyl acetic acid. As found with previous NSAIDs, nabumetone's active metabolite inhibits the cyclooxygenase enzyme and preferentially blocks COX-2 activity
(which is indirectly responsible for the production of inflammation and pain during arthritis). The active metabolite of nabumetone is felt to be the compound primarily responsible for therapeutic effect. Comparatively, the parent drug is a poor inhibitor of COX-2 byproducts, particularly prostaglandins. It may be less nephrotoxic than indomethacin. There
are two known polymorphs of the compound.
|
Naproxen Sodium
https://en.wikipedia.org/wiki/Naproxen
Naproxen, sold under the brand name Aleve among others,
is a nonsteroidal anti-inflammatory drug (NSAID) used to treat pain, menstrual
cramps, inflammatory diseases such as rheumatoid
arthritis, and fever. It is taken orally. It is available in immediate and delayed release formulations.Naproxen works by reversibly inhibiting both the COX-1 and COX-2 enzymes as
a non-selective coxib. This results in the inhibition of prostaglandin synthesis.
Prostaglandins act as signaling molecules in the body, inducing inflammation. Thus, by inhibiting COX-1/2, naproxen induces an anti-inflammatory effect.
|
| Sedatives & Tranquillisers |
Alprazolam
https://en.wikipedia.org/wiki/Alprazolam
Alprazolam, sold under the brand name Xanax,
among others, is a short-acting tranquilizer of the triazolobenzodiazepine (TBZD) class, which are benzodiazepines (BZDs)
fused with a triazole ring. It is most commonly used in short-term management of anxiety
disorders, specifically panic disorder or generalized
anxiety disorder (GAD).
Alprazolam is classed as a high-potency triazolobenzodiazepine: a
benzodiazepine with a triazole ring attached to its structure. As a benzodiazepine, alprazolam produces a variety of therapeutic and adverse effects by binding to the GABAA benzodiazepine
receptor site and modulating its function; GABA receptors are the most prolific inhibitory receptor within the brain.The GABA chemical and receptor system mediates inhibitory or calming effects of alprazolam on the nervous system. Binding of alprazolam to the GABAA receptor,
a chloride ion channel, enhances the effects of GABA, a neurotransmitter. When GABA binds the GABAA receptor the channel opens and chloride enters the cell which makes it more resistant to depolarisation. Therefore, alprazolam has a depressant effect on synaptic transmission to reduce anxiety.
|
Diazepam
ttps://en.wikipedia.org/wiki/Diazepam
Diazepam, first marketed as Valium, is a
medicine of the benzodiazepine family
that typically produces a calming effect. It is commonly used to treat a range of conditions, including anxiety, seizures, alcohol
withdrawal syndrome, benzodiazepine
withdrawal syndrome, muscle
spasms, trouble sleeping, and restless
legs syndrome..enzodiazepines are positive allosteric modulators of the GABA type A receptors (GABAA). The GABAA receptors
are ligand-gated chloride-selective ion channels that are activated by GABA, the major inhibitory neurotransmitter in the brain. Binding of benzodiazepines to this receptor complex promotes the binding of GABA, which in turn increases the total conduction of chloride ions across the neuronal cell membrane. This increased chloride ion influx hyperpolarizes the neuron's membrane potential. As a result, the difference between resting potential and threshold potential is increased and firing is less likely. As a result, the arousal of the cortical and limbic
systems in the central nervous system is reduced.
|
| Tricyclic & Related Antidepressants |
Agomelatine
https://en.wikipedia.org/wiki/Agomelatine
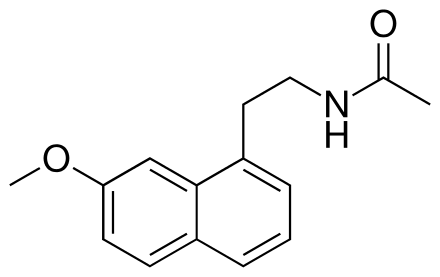
Agomelatine is an atypical
antidepressant used to treat major depressive disorder. One review found that it is as effective as other antidepressants with similar discontinuation rates overall but less
Agomelatine is a melatonin receptor agonist (MT1 (Ki 0.1 nM) and MT2 (Ki =
0.12 nM)) and serotonin 5-HT2C (Ki = 631 nM) and 5-HT2B receptor (Ki =
660 nM) antagonist. Binding studies indicate that it has no effect on monoamine uptake and no affinity for adrenergic, histamine, cholinergic, dopamine,
and benzodiazepine receptors, nor other serotonin receptors.
Agomelatine resynchronizes circadian rhythms in animal models of delayed sleep phase syndrome. By antagonizing 5-HT2C, it disinhibits/increases noradrenaline and dopamine release
specifically in the frontal cortex. Therefore, it is sometimes classified as a norepinephrine–dopamine disinhibitor.
discontinuations due to side effects. Another
review also found it was similarly effective to many other antidepressants.
|
Brexpiprazole
https://en.wikipedia.org/wiki/Brexpiprazole

Brexpiprazole, sold under the brand name Rexulti among
others, is an atypical antipsychotic. It is a dopamine D2 receptor partial
agonist and has been described as a "serotonin–dopamine activity modulator" (SDAM). The drug was approved by the U.S. Food
and Drug Administration (FDA) on July 10, 2015, for the treatment of schizophrenia,
and as an adjunctive treatment for depression. It
has been designed to provide improved efficacy and tolerability (e.g., less akathisia, restlessness and/or insomnia)
over established adjunctive treatments for major depressive disorder (MDD).Brexpiprazole
acts as a partial agonist of the serotonin 5-HT1A receptor and
the dopamine D2 and D3 receptors. Partial
agonists have both blocking properties and stimulating properties at the receptor they bind to. The ratio of blocking activity to stimulating activity determines a portion of its clinical effects.
|
Cinitapride Hydrogen Tartrate
https://en.wikipedia.org/wiki/Cinitapride

Cinitapride (trade names Cintapro, Pemix) is a gastroprokinetic agent and antiulcer agent of the benzamide class which is marketed in India, Mexico, Pakistan and Spain. It
acts as an agonist of the 5-HT1 and 5-HT4 receptors and as an antagonist of
the 5-HT2 receptors.
It is indicated for the treatment of gastrointestinal disorders associated with motility disturbances such as gastroesophageal reflux disease, non-ulcer dyspepsia and delayed gastric emptying
|
| |
Citalopram
https://en.wikipedia.org/wiki/Citalopram

Citalopram,
sold under the brand name Celexa among
others, is an antidepressant of
the selective serotonin reuptake inhibitor (SSRI)
class.[2] It
is used to treat major depressive disorder, obsessive
compulsive disorder, panic
disorder, and social
phobia.Citalopram was first synthesized in 1972 by chemist Klaus Bøgesø and his research group at the pharmaceutical company Lundbeck and
was first marketed in 1989 in Denmark. It was first marketed in the US in 1998.[66] The patent expired
in 2003, allowing other companies to legally produce generic versions.
|
Dothiepin HCl
https://en.wikipedia.org/wiki/Dosulepin

Dosulepin,
also known as dothiepin and
sold under the brand name Prothiaden among
others, is a tricyclic antidepressant (TCA)
which is used in the treatment of depression. Dosulepin
was once the most frequently prescribed antidepressant in the United Kingdom,
but it is no longer widely used due to its relatively high toxicity in overdose without
therapeutic advantages over other TCAs. It acts as a serotonin–norepinephrine reuptake inhibitor (SNRI)
and also has other activities including antihistamine, antiadrenergic, antiserotonergic, anticholinergic,
and sodium channel-blocking effects.Dosulepin
is a transporter blocker of the serotonin
transporter (SERT) and the norepinephrine transporter (NET), thereby acting as an SNRI. It is also an antagonist of
the histamine H1 receptor, α1-adrenergic
receptor, serotonin 5-HT2 receptors,
and muscarinic acetylcholine receptors (mACh), as well as a blocker of voltage-gated
sodium channels (VGSCs). The antidepressant effects of dosulepin are thought to be due to inhibition of the reuptake of norepinephrine and
possibly also of serotonin..
|
Escitalopram
https://en.wikipedia.org/wiki/Escitalopram

Escitalopram,
sold under the brand names Cipralex and Lexapro,
among others, is an antidepressant of
the selective serotonin reuptake inhibitor (SSRI)
class. Escitalopram is
mainly used to treat major depressive disorder or generalized
anxiety disorder. It
is taken by mouth.
Escitalopram increases intrasynaptic levels of the neurotransmitter serotonin by
blocking the reuptake of
the neurotransmitter into the presynaptic neuron. Of the SSRIs currently available, escitalopram has the highest selectivity for
the serotonin transporter (SERT)
compared to the norepinephrine transporter (NET),
making the side-effect profile relatively mild in comparison to less-selective SSRIs.
|
Fluoxetine Hydrochloride
https://en.wikipedia.org/wiki/Fluoxetine

Fluoxetine,
sold under the brand names Prozac and Sarafem among
others, is an antidepressant of
the selective serotonin reuptake inhibitor (SSRI)
class. It is used for
the treatment of major depressive disorder, obsessive–compulsive
disorder (OCD), bulimia
nervosa, panic
disorder, and premenstrual
dysphoric disorder. It
may decrease the risk of suicide in those over the age of 65. It
has also been used to treat premature ejaculation.Fluoxetine
elicits antidepressant effect by inhibiting serotonin re-uptake in the synapse by binding to the re-uptake pump on the neuronal membrane to
increase its availability and enhance neurotransmission. Norfluoxetine and desmethylfluoxetine are metabolites of fluoxetine and also act as serotonin re-uptake inhibitors, so increase the duration of action of the drug.
|
Milnacipran
https://en.wikipedia.org/wiki/Milnacipran
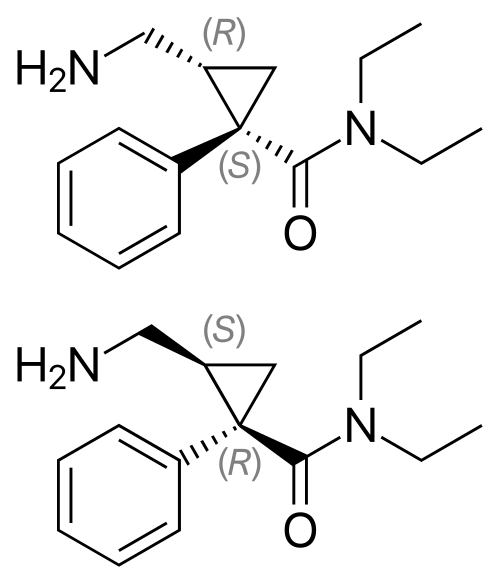
Milnacipran (trade
names Ixel, Savella, Dalcipran, Toledomin)
is a serotonin–norepinephrine reuptake inhibitor (SNRI)
used in the clinical treatment of fibromyalgia.
It is not approved for the clinical treatment of major depressive disorder in
the US, but it is in other countries.
Milnacipran inhibits the reuptake of serotonin and norepinephrine in an approximately 1:3 ratio, respectively. Milnacipran exerts no significant actions on H1, α1, D1, D2,
and mACh receptors, nor on benzodiazepine and opioid binding
sites.
Recently, levomilnacipran, the levorotatory enantiomer of milnacipran, has been found to act as an inhibitor of beta-site
amyloid precursor protein cleaving enzyme-1 (BACE-1), which is responsible for β-amyloid plaque formation, and hence may be a potentially useful drug in the treatment of Alzheimer's disease Other BACE-1 inhibitors, such as CTS-21166 (ASP1720), MK-8931,
and AZD3293 are currently in clinical trials for the treatment of Alzheimer's disease.
|
Nortriptyline
https://en.wikipedia.org/wiki/Nortriptyline

Nortriptyline,
sold under the brand name Pamelor, among
others, is a medication used to treat depression, neuropathic
pain, attention
deficit hyperactivity disorder (ADHD), stopping
smoking and anxiety.
Nortriptyline is an active metabolite of amitriptyline by demethylation in the liver. Its pharmacologic profile is as the table to the right shows (inhibition or antagonism of all sites).
Chemically, it is a secondary amine dibenzocycloheptene and pharmacologically it is classed as a first-generation antidepressant.
|
Sertraline HCl
https://en.wikipedia.org/wiki/Sertraline
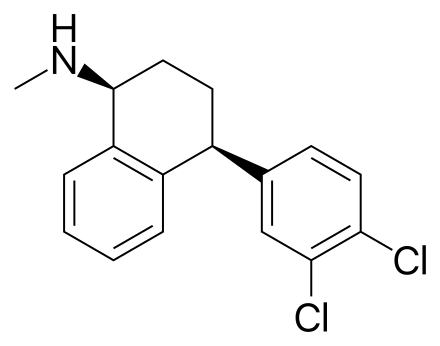
Sertraline,
sold under the brand name Zoloft among
others, is an antidepressant of
the selective serotonin reuptake inhibitor (SSRI)
class. It is used to
treat major depressive disorder, obsessive–compulsive
disorder, panic
disorder, post-traumatic
stress disorder, premenstrual
dysphoric disorder, and social
anxiety disorder.Sertraline acts as a potent serotonin
reuptake inhibitor (SRI), with an affinity (Ki)
for the serotonin transporter (SERT) of 0.4 nM and an IC50 value
of 2.8 nM, according to a couple of studies. It is highly selective in its inhibition of serotonin reuptake. By
inhibiting the reuptake of serotonin, sertraline increases extracellular levels of serotonin and thereby increases serotonergic neurotransmission in
the brain. It is this action that is thought to be responsible for the antidepressant, anxiolytic,
and antiobsessional effects of sertraline.
|
Imipramine
https://en.wikipedia.org/wiki/Imipramine

Imipramine, sold under the brand name Tofranil,
among others, is a tricyclic antidepressant (TCA)
mainly used in the treatment of depression. It is also effective in treating anxiety and panic
disorder. The drug is also used to treat bedwetting.. It
was the first TCA to be marketed. Imipramine and the other TCAs have decreased in use in recent decades, due to the introduction of the selective serotonin reuptake inhibitors (SSRIs),
which have fewer side effects and are safer in overdose.
|
Trimipramine Maleate
https://en.wikipedia.org/wiki/Imipramine
Imipramine,
sold under the brand name Tofranil, among others, is a tricyclic
antidepressant (TCA) mainly
used in the treatment of depression.
It is also effective in treating anxiety and panic
disorder. The drug is also used to treat bedwetting..The
mechanisms of imipramine's actions include, but are not limited to, effects on:
D2 receptors Imipramine, and its metabolite desipramine, have no appreciable affinity for the dopamine transporter (Ki = 8,500 and >10,000 nM, respectively)...............
|
Vilazodone
https://en.wikipedia.org/wiki/Vilazodone

Vilazodone,
sold under the brand name Viibryd among
others, is a medication used to treat major depressive disorder.[1] While
it was being studied for generalized anxiety disorder,
such research had stopped as of 2017..Vilazodone acts as a serotonin
reuptake inhibitor (IC50 =
2.1 nM; Ki =
0.1 nM) and 5-HT1A receptor partial
agonist (IC50 =
0.2 nM; IA = ~60–70%).It has negligible affinity for
other serotonin receptors such
as 5-HT1D, 5-HT2A,
and 5-HT2C. It
also exhibits clinically unimportant inhibitory activity at the norepinephrine and dopamine transporters (Ki =
56 nM for NET and
37 nM for DAT).
|
Amitriptyline HCl
https://en.wikipedia.org/wiki/Amitriptyline
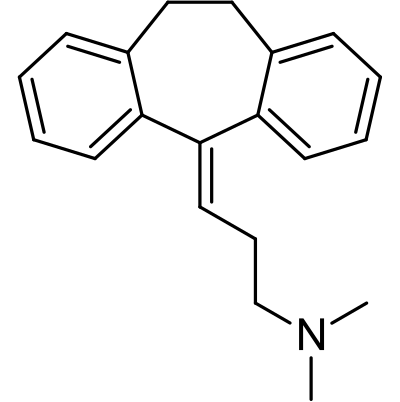
Amitriptyline inhibits neuronal reuptake of serotonin and noradrenaline from the synapse in the central nervous system; this increases their availability in the synapse to cause neurotransmission on the post-synaptic neurone.Amitriptyline is metabolised by cytochrome P450 enzymes in the liver to nortriptyline, which also acts as a noradrenaline reuptake inhibitor; this potentiates the antidepressant effects of amitriptyline. |
|
| Contraceptive agents |
| Oral Contraceptives |
Ulipristal Acetate
https://en.wikipedia.org/wiki/Ulipristal_acetate

Ulipristal acetate, sold under the brand name Ella among
others, is a medication used for emergency contraception (birth
control) and uterine fibroids.[1][2][3] As
emergency contraception it should be used within 120 hours of sex. For
fibroids it may be taken for up to six months. It
is taken by mouth.As an SPRM, ulipristal acetate has partial agonistic as well as antagonistic effects
on the progesterone receptor. Ulipristal acetate exhibits similar potency to antagonize progesterone
receptor as mifepristone in vitro. It also binds to
the androgen receptor and the glucocorticoid
receptor, but is only a weak antiandrogen and antiglucocorticoid relative
to flutamide and mifepristone,
respectively. |
|
|
| Dermatologicals |
| Anti- Infective Skin Preparations |
Acyclovir
https://en.wikipedia.org/wiki/Aciclovir
Aciclovir (ACV),
also known as acyclovir, is an antiviral
medication. It
is primarily used for the treatment of herpes simplex virus infections, chickenpox,
and shingles.Other
uses include prevention of cytomegalovirus infections
following transplant and severe complications of Epstein-Barr virus infection.Aciclovir
is converted by viral thymidine kinase to aciclovir monophosphate, which is then converted by host cell kinases to
aciclovir triphosphate (ACV-TP). ACV-TP, in turn, competitively inhibits and inactivates HSV-specified DNA
polymerases preventing further viral DNA synthesis without affecting the normal cellular processes.
|
Polymyxin B Sulfate
https://en.wikipedia.org/wiki/Polymyxin_B
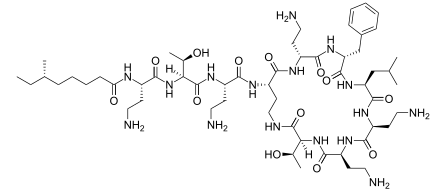
Polymyxin B,
sold under the brand name Poly-Rx among
others, is an antibiotic used
to treat meningitis, pneumonia, sepsis,
and urinary tract infections. While
it is useful for many Gram negative infections,
it is not useful for Gram positive infections..
- Alters bacterial outer membrane permeability by binding to a negatively charged site in the lipopolysaccharide layer, which has an electrostatic attraction for the positively charged amino groups in the cyclic peptide portion (this site normally is a binding site for calcium and magnesium counter ions); the result is a destabilized outer membrane
- Fatty acid portion dissolves in hydrophobic region of cytoplasmic membrane and disrupts membrane integrity
- Leakage of cellular molecules, inhibition of cellular respiration
- Binds and inactivates
endotoxin
Relative absence of selective toxicity: nonspecific for cell membranes of any type, highly toxic.
|
Silver Sulfadiazine
https://en.wikipedia.org/wiki/Silver_sulfadiazine

Silver sulfadiazine,
sold under the brand Silvadene among
others, is a topical antibiotic used
in partial thickness and full thickness burns to
prevent infection. Tentative
evidence has found other antibiotics to be more effective and therefore it is no longer generally recommended.The chemical is poorly soluble, and has only very limited penetration through intact skin. However, contact with body fluids produces free sulfadiazine which can then be systemically absorbed and distributed; it undergoes glucuronidation in
the liver and is also excreted unaltered in urine. Only
when applied to large-area (especially second- and third-degree) burns or other lesions is absorption into the body a problem. |
Selenium Sulphide
https://en.wikipedia.org/wiki/Selenium_disulfide
Selenium disulfide, also known as selenium
sulfide, is a chemical compound and medication used to treat pityriasis
versicolor, seborrhoeic
dermatitis, and dandruff. It
is applied to the affected area as a lotion or shampoo. Dandruff
frequently returns if treatment is stopped.Selenium disulfide is sold as an antifungal agent in shampoos for
the treatment of dandruff and seborrheic
dermatitis associated in the scalp with fungi of genus Malassezia. In the United
States, a 1% strength is available over-the-counter, and a 2.5% strength is also available with a prescription.
In Canada, the 2.5% strength is available over-the-counter. At the 2.5% strength, selenium disulfide is also used on the body to treat Tinea versicolor, a type of fungal skin
infection caused by a different species of Malassezia. It has been suggested to be effective as a treatment for hyperkeratosis..
|
| Antipruritic |
Crotamiton
https://en.wikipedia.org/wiki/Crotamiton

Crotamiton is
a drug that is used both as a scabicidal (for treating scabies)
and as a general antipruritic (anti-itching
drug). It is a prescription, lotion-based medicine that is applied to the whole body to get rid of the scabies parasite that
burrows under the skin and
causes itching.
Crotamiton is toxic to the scabies mite, The
mechanism of action of crotamiton as a general antipruritic was reported, in which crotamiton inhibits TRPV4 (transient receptor potential vanilloid 4) channel that is expressed in the skin,
primary sensory neurons, and so on.
|
| Antipsoriasis |
Acitretin
https://en.wikipedia.org/wiki/Acitretin

Acitretin (trade names Soriatane and Neotigason) is a second-generation retinoid. It is taken orally, and is typically used for psoriasis.
Acitretin is an oral retinoid used in the treatment of severe resistant psoriasis. Because of the potential for problems and severe side effects it is generally used in only very severe cases of psoriasis that have been unresponsive to other treatments. It binds to nuclear receptors that regulates gene transcription. They induce keratinocyte differentiation and reduce epidermal hyperplasia, leading to the slowing of cell reproduction.
|
Dimethyl Fumarate
https://en.wikipedia.org/wiki/Dimethyl_fumarate

Dimethyl fumarate (DMF)
is the methyl ester of fumaric
acid and is named after the
earth smoke plant (Fumaria officinalis). DMF
combined with three other fumaric acid esters
(FAEs) is solely licensed in Germany as an oral therapy for psoriasis (trade
name Fumaderm). Since
2013, it has been approved by the U.S. Food and Drug Administration as
a treatment option for adults with relapsing multiple sclerosis (trade
name Tecfidera).DMF has also been applied as a biocide in
furniture or shoes to prevent growths of mold during storage or transport in a humid climates. However, due to cases of allergic reactions after skin contact, DMF-containing consumer products are no longer authorised to be manufactured (since 1998) or imported (since 2009) in the European
Union.. |
Methoxsalen
https://en.wikipedia.org/wiki/Methoxsalen

Methoxsalen (also
called xanthotoxin), sold under the brand name Oxsoralen among
others, is a drug used
to treat psoriasis, eczema, vitiligo,
and some cutaneous lymphomas in
conjunction with exposing the skin to UVA light from lamps or sunlight. Methoxsalen modifies the way skin cells receive the UVA radiation,
allegedly clearing up the disease. The dosage comes in 10 mg tablets, which are taken in the amount of 30 mg 75 minutes before a PUVA (psoralen + UVA)
light treatment. Levels of individual patient PUVA exposure were originally determined using the Fitzpatrick scale.
The scale was developed after patients demonstrated symptoms of phototoxicity after
oral ingestion of Methoxsalen followed by PUVA therapy. Chemically, methoxsalen belongs to a class of organic natural molecules known as furanocoumarins.
They consist of coumarin annulated with furan.
|
| Corticosteroid Responsive |
Methylprednisolone & Salts
https://en.wikipedia.org/wiki/Methylprednisolone

Methylprednisolone,
sold under the brand name Medrol among
others, is a corticosteroid medication
used to suppress the immune system and
decrease inflammation. Conditions
in which it is used include skin diseases, rheumatic disorders,
allergies, asthma, croup, COPD,
certain cancers, multiple
sclerosis, and as add-on therapy for tuberculosis or
radiculopathyUnbound glucocorticoids cross cell membranes and bind with high affinity to specific cytoplasmic receptors, modifying transcription and protein synthesis. By this mechanism, glucocorticoids can inhibit leukocyte infiltration at the site of inflammation, interfere with mediators of inflammatory response, and suppress humoral immune responses. The anti-inflammatory actions of corticosteroids are thought to involve phospholipase A2 inhibitory proteins, lipocortins, which control the
biosynthesis of potent mediators of inflammation such as prostaglandins and leukotrienes.
|
| Drugs for Acne Vulgaris |
Adapalene
https://en.wikipedia.org/wiki/Adapalene

Adapalene is
a third-generation topical retinoid primarily
used in the treatment of mild-moderate acne,
and is also used off-label to
treat keratosis pilaris as
well as other skin conditions.[1] It
is effective against acne conditions where comedones are
predominant.Unlike the retinoid tretinoin (Retin-A), adapalene has also been shown to retain its efficacy when applied at the same time as benzoyl
peroxide due to its more stable chemical structure.[14] Furthermore, photodegradation of
the molecule is less of a concern in comparison to tretinoin and tazarotene.
|
| Melanizing Agents |
Trioxsalen
https://en.wikipedia.org/wiki/Trioxsalen

Trioxsalen (trimethylpsoralen, trioxysalen (INN)
or Trisoralen) is a furanocoumarin and
a psoralen derivative.
It is obtained from several plants, mainly Psoralea corylifolia.
Like other psoralens it causes photosensitization of
the skin. It is administered either topically or orally in
conjunction with UV-A (the
least damaging form of ultraviolet light)
for phototherapy treatment
of vitiligo and
hand eczema. After photoactivation it
creates interstrand cross-links in
DNA, which can cause programmed cell death unless
repaired by cellular mechanisms. In research it can be conjugated to dyes for confocal microscopy and
used to visualize sites of DNA damage. The
compound is also being explored for development of antisense oligonucleotides that
can be cross-linked specifically to a mutant mRNA sequence
without affecting normal transcripts differing at even a single base pair.
|
| Topical Antifungals & Anti-Parasitics |
Sertaconazole Nitrate
https://en.wikipedia.org/wiki/Sertaconazole

Sertaconazole, sold under the brand names Ertaczo, Dermofix, Konzert, and Zalain, is an antifungal medication of the imidazole class. It is available as a cream to treat skin infections
such as athlete's foot.
It is also available in a vaginal tablet form. The most popular of these is Gyno-Dermofix.
Like other imidazole antifungals, sertaconazole blocks the synthesis of ergosterol by inhibiting the 14α-demethylase enzyme. Ergosterol is a critical component of the fungal cell membrane. Inhibition of ergosterol synthesis prevents fungal cells from multiplying and impairs hyphae growth.
|
Terbinafine
https://en.wikipedia.org/wiki/Terbinafine

Terbinafine,
sold under the brand name Lamisil among
others, is an antifungal medication used
to treat pityriasis versicolor, fungal
nail infections, and ringworm including jock
itch and athlete's
foot.
Like other allylamines, terbinafine inhibits ergosterol synthesis by inhibiting squalene epoxidase, an enzyme that catalyzes the conversion of squalene to lanosterol. In fungi, lanosterol is then converted to ergosterol;
in humans, lanosterol becomes cholesterol. However, there is sufficient genetic divergence between fungal and human squalene epoxidases that terbinafine preferentially binds fungal squalene epoxidase, making it selective for inhibiting ergosterol production in fungi without significantly affecting cholesterol production in humans. This is thought to change cell membrane permeability, causing fungal cell lysis.
Terbinafine is highly lipophilic and tends to accumulate in hair, skin, nails, and fatty tissue.
|
| Topical Steroids |
Betamethasone
https://en.wikipedia.org/wiki/Betamethasone

Betamethasone is
a steroid medication. It
is used for a number of diseases including rheumatic disorders such
as rheumatoid arthritis and systemic
lupus erythematosus, skin diseases such as dermatitis and psoriasis,
allergic conditions such as asthma and angioedema, preterm
labor to speed the
development of the baby's lungs, Crohn's disease,
cancers such as leukemia,
and along with fludrocortisone for adrenocortical
insufficiency, among others.
|
Fluticasone Propionate
https://en.wikipedia.org/wiki/Fluticasone_propionate

Fluticasone propionate,
sold under the brand names Flovent and Flonase among
others, is a steroid medication.[6] When
inhaled it is used for the long term management of asthma and COPD.[6] In
the nose it is used for hay fever and nasal
polyps.[7][8] It
can also be used for mouth ulcers..Fluticasone
propionate is a highly selective agonist at
the glucocorticoid receptor with
negligible activity at androgen, estrogen,
or mineralocorticoid receptors,
thereby producing anti-inflammatory and vasoconstriction effects.
It has been shown to have a wide range of inhibitory effects on multiple cell types (e.g. mast cell, eosinophil, neutrophil, macrophages,
and lymphocytes)
and mediators (e.g. histamine, eicosanoids, leukotrienes,
and cytokines)
involved in inflammation.
Fluticasone propionate is stated to exert a topical effect on the lungs without significant systemic effects at usual doses, due to its low systemic bioavailability.
|
Mometasone Furoate
https://en.wikipedia.org/wiki/Mometasone

Mometasone,
also known as mometasone furoate, is a steroid medication
used to treat certain skin conditions, hay
fever, and asthma. Specifically
it is used to prevent rather than treat asthma attacks. It
can be applied to the skin, inhaled, or used in the nose. Mometasone furoate,
not mometasone is used in medical products.
Mometasone, like other corticosteroids, possesses anti-inflammatory, antipruritic, and vasoconstrictive properties. For allergies, corticosteroids reduce the allergic reactions in various types of cells (mastocytes and eosinophils) that are responsible for allergic reactions. Mometasone and other corticosteroids circulate in the blood easily, crossing cellular membranes and binding with cytoplasmic receptors, resulting in the transcription and synthesis of proteins. It also inhibits the actions of the enzyme cytochrome P450 2C8 which participates in the activity of monooxygenase.
The inflammation is reduced in decreasing the liberation of hydrolase acids of leukocytes, the prevention of the accumulation macrophages in the sites of inflammation, the interference with adhesion of leukocytes to capillary walls, the reduction of the permeability of the capillary membranes and consequently edema, the reduction of complementary components, inhibition of histamine and kinin liberation, and interference with scar tissue formation.
|
|
| |
|
| Opthalmic |
| Anti- Infective Eye Preparations |
Acyclovir
https://en.wikipedia.org/wiki/Aciclovir
Aciclovir (ACV),
also known as acyclovir, is an antiviral
medication. It
is primarily used for the treatment of herpes simplex virus infections, chickenpox,
and shingles. Other
uses include prevention of cytomegalovirus infections
following transplant and severe complications of Epstein-Barr virus infection.Aciclovir
is converted by viral thymidine kinase to aciclovir monophosphate, which is then converted by host cell kinases to
aciclovir triphosphate (ACV-TP). ACV-TP, in turn, competitively inhibits and inactivates HSV-specified DNA
polymerases preventing further viral DNA synthesis without affecting the normal cellular processes.
|
Fluconazole
https://en.wikipedia.org/wiki/Fluconazole
Fluconazole is
an antifungal medication used
for a number of fungal infections. This
includes candidiasis, blastomycosis, coccidiodomycosis, cryptococcosis, histoplasmosis, dermatophytosis,
and pityriasis versicolor. It
is also used to prevent candidiasis in those who are at high risk such as following organ transplantation,
low birth weight babies, and those with low blood neutrophil counts.Like
other imidazole- and triazole-class
antifungals, fluconazole inhibits the fungal cytochrome P450 enzyme 14α-demethylase.
Mammalian demethylase activity is much less sensitive to fluconazole than fungal demethylase. This inhibition prevents the conversion of lanosterol to ergosterol,
an essential component of the fungal cytoplasmic membrane, and subsequent accumulation of 14α-methyl sterols.[22] Fluconazole
is primarily fungistatic; however, it may be fungicidal against certain organisms in a dose-dependent manner, specifically Cryptococcus.
|
| Anti- Inflammatory & Anti Allergic Preparations |
Ketorolac Tromethamine
https://en.wikipedia.org/wiki/Ketorolac

Ketorolac,
sold under the brand name Toradol among
others, is a nonsteroidal anti-inflammatory drug (NSAID)
used to treat pain..Chemically
ketorolac functions as a carboxylic acid derivative serving non-selectively to block the prostaglandin synthesis by inhibition of prostaglandin G/H synthesis one and two. Prostaglandin functions in the body as a messenger for contraction/relaxation of smooth muscle and modulation of inflammation. Resultant, inhibition of prostaglandin synthesis prevents inflammation. The
primary mechanism of action responsible
for ketorolac's anti-inflammatory, antipyretic,
and analgesic effects is the inhibition of prostaglandin synthesis by competitive blocking of the enzyme cyclooxygenase (COX).
Ketorolac is a non-selective COX inhibitor. It
is considered a first-generation NSAID.
|
Sodium Cromoglycate
https://en.wikipedia.org/wiki/Cromoglicic_acid
Cromoglicic acid (INN)
(also referred to as cromolyn (USAN), cromoglycate (former BAN),
or cromoglicate) is traditionally described as a mast
cell stabilizer, and is commonly marketed as the sodium salt sodium
cromoglicate or cromolyn
sodium. This drug prevents the release of inflammatory chemicals
such as histamine from mast
cells.Cromolyn works because it prevents the release of mediators that would normally attract inflammatory cells and because it stabilizes the inflammatory cells. MCT mast cells found in the mucosa are stabilised." Nedocromil is
another mast cell stabilizer that also works in controlling asthma. The underlying mechanism of action is not fully understood; for while cromoglicate stabilizes mast cells, this mechanism is probably not why it works in asthma.
|
| |
|
| Glaucoma |
Bimatoprost
https://en.wikipedia.org/wiki/Bimatoprost

Bimatoprost,
sold under the trade name Lumigan among
others, is a medication used to treat high pressure inside the eye including glaucoma. Specifically
it is used for open angle glaucoma when
other agents are not sufficient. It
may also be used to increase the size of the eyelashes.Bimatoprost
is a structural analog of prostaglandin
F2α (PGF2α).
Like other PGF2α analogs such as travoprost, latanoprost and tafluprost,
it increases the outflow of aqueous fluid from the eye and lowers intraocular pressure. However, in contrast to these it does not act on the prostaglandin F receptor, nor on any other known prostaglandin receptor. It is thought that bimatoprost mimics the human body's own prostamides (which
are chemically similar), a class of substances related to prostaglandins, but with an unknown mechanism of action. No prostamide receptor has been identified as of 2015; the search is ongoing.As of 2019 it was thought that bimatoprost worked via the trabecular meshwork and uveoscleral pathways..
|
Dorzolamide
https://en.wikipedia.org/wiki/Dorzolamide

Dorzolamide,
sold under the brand name Trusopt among
others, is medications used to treat high pressure inside the eye including glaucoma.It
lowers IOP by
about 20%. Carbonic Anhydrase can convert H2CO3 into HCO3 (bicarbonate) and H+. The H+ is then exchanged for sodium (Na) which allows you to make aqueous humor. By blocking carbonic anhydrase, the Na/H exchanger can't work, which will decrease Na in the cell and prevent aqueous humor production.
|
Timolol Maleate
https://en.wikipedia.org/wiki/Timolol

Timolol is
a medication used either by mouth or as eye drops. As
eye drops it is used to treat increased pressure inside the eye such
as in ocular hypertension and glaucoma. By
mouth it is used for high blood pressure, chest
pain due to insufficient blood flow to the heart, to prevent further complications after a heart
attack, and to prevent migraines.Beta
blockers are competitive antagonists that
block the receptor sites for the endogenous catecholamines epinephrine (adrenaline)
and norepinephrine (noradrenaline) on adrenergic
beta receptors, of the sympathetic nervous system, which mediates the fight-or-flight
response. Some block activation of all types of β-adrenergic receptors and others are selective for one of the three known types of beta receptors, designated β1,
β2 and β3 receptors β1-adrenergic
receptors are
located mainly in the heart and in the kidneys.
|
| Mydriatics & Cycloplegics |
Phenylephrine
https://en.wikipedia.org/wiki/Phenylephrine
Phenylephrine is
a medication primarily used as a decongestant,
to dilate the pupil,
to increase blood pressure,
and to relieve hemorrhoids. While
marketed as a decongestant, taken by mouth at recommended doses it is of unclear benefit for hay fever.
Phenylephrine is a sympathomimetic drug, which means that it mimics the actions of epinephrine (commonly known as adrenaline) or norepinephrine. Phenylephrine selectively binds to alpha-1 receptors which cause blood vessels to constrict.
Whereas pseudoephedrine causes both vasoconstriction and increase of mucociliary clearance through its nonspecific adrenergic activity, phenylephrine's selective α-adrenergic agonism causes vasoconstriction alone, creating a difference in their methods of action.
|
| |
|
| Genito-urinary tract |
| Androgen & Anabolic Steroid (AAS) |
Stanozolol
https://en.wikipedia.org/wiki/Stanozolol
Stanozolol (abbrev. Stz),
sold under many brand names, is an androgen and anabolic
steroid (AAS) medication
derived from dihydrotestosterone (DHT).
It is used to treat hereditary angioedema..Stanozolol
is one of the AAS commonly used as performance-enhancing drugs and
is banned from use in sports competition under the auspices of the International Association of Athletics Federations (IAAF)
and many other sporting bodies. Additionally, stanozolol has been used in US horse racing.As
an AAS, stanozolol is an agonist of the androgen
receptor (AR), similarly to androgens like testosterone and
DHT. Its affinity for the androgen receptor is about 22% of that of dihydrotestosterone. Stanozolol
is not a substrate for 5α-reductase as
it is already 5α-reduced, and so is not potentiated in so-called "androgenic" tissues like the skin, hair
follicles, and prostate gland. This results in a greater ratio of anabolic to androgenic activity
compared to testosterone. In addition, due to its 5α-reduced nature, stanozolol is non-aromatizable,
and hence has no propensity for producing estrogenic effects such as gynecomastia or fluid
retention. Stanozolol also does not possess any progestogenic activity of significance. Because of the presence of its 17α-methyl group, the metabolism of
stanozolol is sterically hindered, resulting in it being orally
active, although also hepatotoxic.
|
| Erectile Dysfunction |
Sildenafil Citrate
https://en.wikipedia.org/wiki/Sildenafil

Sildenafil, sold under the brand name Viagra among
others, is a medication used
to treat erectile dysfunction and pulmonary
arterial hypertension. It
is unclear if it is effective for treating sexual dysfunction in women.Sildenafil protects cyclic guanosine monophosphate (cGMP) from degradation by cGMP-specific
phosphodiesterase type 5 (PDE5) in the corpus cavernosum. Nitric
oxide (NO) in the corpus cavernosum of the penis binds to guanylate cyclase receptors, which results in increased levels of cGMP, leading to smooth
muscle relaxation (vasodilation) of the intimal cushions of the helicine
arteries. This smooth muscle relaxation leads to vasodilation and increased inflow of blood into the spongy tissue of the penis, causing an erection. Robert F. Furchgott, Ferid
Murad, and Louis Ignarro won the Nobel
Prize in Physiology or Medicine in 1998 for their independent study of the metabolic pathway of nitric oxide in smooth muscle vasodilation.
|
| Hyperphosphatemia |
Sevelamer HCl / Carbonate
https://en.wikipedia.org/wiki/Sevelamer

Sevelamer (rINN)
( or )
is a phosphate binding drug
used to treat hyperphosphatemia in
patients with chronic kidney disease.
When taken with meals, it binds to dietary phosphate and prevents its absorption.Sevelamer consists of polyallylamine that
is crosslinked with epichlorohydrin. The
marketed form sevelamer hydrochloride is a partial hydrochloride salt being present as approximately 40% amine hydrochloride and
60% sevelamer base. The amine groups of sevelamer become partially protonated in the intestine and interact with phosphate ions through ionic and hydrogen
bonding..
|
Sucroferric Oxyhydroxide
https://en.wikipedia.org/wiki/Sucroferric_oxyhydroxide

Sucroferric oxyhydroxide (trade name Velphoro) is a non-calcium, iron-based phosphate binder used for the control of serum phosphorus levels in adult patients with chronic kidney disease (CKD) on haemodialysis (HD) or peritoneal dialysis (PD). It is used in form of chewable tablets.
Sucroferric oxyhydroxide is also known as a mixture of polynuclear iron(III)-oxyhydroxide, sucrose and starches.
|
| Intravenous Pyelogram (IVP) |
Diatrizoate Sodium Diatrizoic Acid
https://en.wikipedia.org/wiki/Diatrizoate

Meglumine is an amino
sugar derived from glucose. It is often used as an excipient in
pharmaceuticals and in conjunction with iodinated compounds in contrast media such as diatrizoate
meglumine, iothalamate
meglumine and iodipamide meglumine..
|
Meglumine
https://en.wikipedia.org/wiki/Meglumine

Meglumine is an amino
sugar derived from glucose. It is often used as an excipient in
pharmaceuticals and in conjunction with iodinated compounds in contrast media such as diatrizoate
meglumine, iothalamate
meglumine and iodipamide meglumine..
|
| Urinary Tract Analgesics & Antispasmodics |
Adiphenine HCl
https://en.wikipedia.org/wiki/Adiphenine

Adiphenine is
an inhibitor of nicotinic receptors.
|
Drotaverine HCl
https://en.wikipedia.org/wiki/Drotaverine

Drotaverine (INN, also known as drotaverin) is an antispasmodic drug, used to enhance cervical dilation during childbirth.
It is structurally related to papaverine, is a selective inhibitor of phosphodiesterase 4, and has no anticholinergic effects.
|
Phenazopyridine
https://en.wikipedia.org/wiki/Phenazopyridine

Phenazopyridine is
a medication which, when excreted into the urine,
has a local analgesic effect.
It is often used to help with the pain, irritation,
or urgency caused
by urinary tract infections, surgery,
or injury to the urinary tract.Phenazopyridine's mechanism
of action is
not well known, and only basic information on its interaction with the body is available. It is known that the chemical has a direct topical analgesic effect on the mucosa lining of the urinary tract. It is rapidly excreted by the kidneys directly into the urine.[20] Hydroxylation is
the major form of metabolism in humans,[20] and the azo bond
is usually not cleaved. On the order of 65% of an oral dose will be secreted directly into the urine chemically unchanged.
|
Trospium Chloride
https://en.wikipedia.org/wiki/Trospium_chloride
Trospium chloride is
used to treat overactive bladder..Chemically
it is a quaternary ammonium cation which
causes it to stay in periphery rather than crossing the blood-brain barrier.[5] It
works by causing the smooth muscle in
the bladder to relax.Trospium chloride is a muscarinic antagonist. Trospium chloride blocks the effect of acetylcholine on muscarinic receptors
organs that are responsive to the compounds, including the bladder.[2] Its parasympatholytic action
relaxes the smooth muscle in the bladder.[3] Receptor assays showed that trospium chloride has negligible affinity for nicotinic receptors as compared to muscarinic receptors at concentrations obtained from therapeutic doses.
|
| Urinary Tract Infections |
Fosfomycin Trometamol
https://en.wikipedia.org/wiki/Fosfomycin

Fosfomycin,
sold under the brand name Monurol among
others, is an antibiotic primarily
used to treat bladder infections. It
is not recommended for kidney infections.Fosfomycin
is bactericidal and inhibits bacterial cell wall biogenesis by inactivating the enzyme UDP-N-acetylglucosamine-3-enolpyruvyltransferase,
also known as MurA. This enzyme catalyzes the committed step in peptidoglycan biosynthesis,
namely the ligation of phosphoenolpyruvate (PEP) to the 3'-hydroxyl group of UDP-N-acetylglucosamine.
This pyruvate moiety provides the linker that bridges the glycan and peptide portion of peptidoglycan. Fosfomycin is a PEP analog that inhibits MurA by alkylating an active site cysteine residue
(Cys 115 in the Escherichia coli enzyme)..
|
| Uterine Stimulants |
Ritodrine Hydrochloride
https://en.wikipedia.org/wiki/Ritodrine

Ritodrine (trade
name Yutopar) is a tocolytic drug used
to stop premature labor.This
drug has been removed from the US market, according to FDA Orange Book.Ritodrine
is a short-acting β2 adrenoreceptor
agonist — a class of medication used for smooth muscle relaxation (other similar drugs are used in asthma or other pulmonary diseases such as salbutamol (albuterol)).
Since ritodrine has a bulky N-substituent, it has high β2 selectivity. Also, the 4-hydroxy
group on the benzene ring is important for activity as it is needed to form hydrogen bonds. However, the 4-hydroxy group makes it susceptible to metabolism by catechol-O-methyl
transferase (COMT).
Since it is β2-selective it is used for premature labor..
|
| |
|
| Hormones |
| Corticosteroids & Related Drugs |
Deflazacort
https://en.wikipedia.org/wiki/Deflazacort
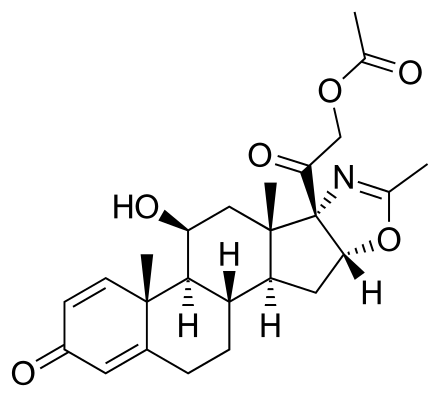
Deflazacort (trade
name Calcort among
others) is a glucocorticoid used
as an anti-inflammatory and immunosuppressant.Deflazacort
is an inactive prodrug which
is metabolized rapidly to the active drug 21-desacetyldeflazacort.
|
| Hyper & Hypoglycaemics |
Glibenclamide
https://en.wikipedia.org/wiki/Glibenclamide

Glibenclamide,
also known as glyburide, is a medication used to treat diabetes
mellitus type 2. It
is recommended that it be taken together with diet and exercise.[1] It
may be used with other antidiabetic medication.[1] It
is not recommended for use by itself in diabetes mellitus type 1.
The medication works by binding to and inhibiting the ATP-sensitive potassium channels (KATP) inhibitory regulatory subunit sulfonylurea receptor 1 (SUR1)[10] in pancreatic beta
cells. This inhibition causes cell membrane depolarization, opening voltage-dependent calcium channels. This results in an increase in intracellular calcium in the pancreatic beta cell and subsequent stimulation of insulin release.
After a cerebral ischemic insult, the blood–brain barrier is broken and glibenclamide can reach the central nervous system. Glibenclamide has been shown to bind more efficiently to the ischemic hemisphere.
|
Glimepiride
https://en.wikipedia.org/wiki/Glimepiride

Glimepiride, sold under the trade name Amaryl among
others, is a medication used to treat diabetes mellitus type 2. It
is less preferred than metformin. Use
is recommended together with diet and exercise.
Like all sulfonylureas, glimepiride acts as an insulin secretagogue. It lowers blood sugar by stimulating the release of insulin by pancreatic beta cells and by inducing increased activity of intracellular insulin receptors.
Not all secondary sulfonylureas have the same risk of hypoglycemia. Glibenclamide (glyburide) is associated with an incidence of hypoglycemia of up to 20–30%, compared to as low as 2% to 4% with glimepiride. Glibenclamide also interferes with the normal homeostatic suppression of insulin secretion in reaction to hypoglycemia, whereas glimepiride does not. Also, glibenclamide diminishes glucagon secretion in reaction to hypoglycemia, whereas glimepiride does not.
|
| Thyroid Hormones |
Levothyroxine Sodium
https://en.wikipedia.org/wiki/Levothyroxine

Levothyroxine,
also known as L-thyroxine, is a manufactured form of the thyroid
hormone thyroxine (T4). It
is used to treat thyroid hormone deficiency,
including the severe form known as myxedema coma. It
may also be used to treat and prevent certain types of thyroid tumors. It
is not indicated for weight loss.Levothyroxine
is a synthetic form of thyroxine (T4),
an endogenous hormone secreted by the thyroid gland, which is converted to its active metabolite, L-triiodothyronine (T3). T4 and
T3 bind to thyroid receptor proteins in the cell
nucleus and cause metabolic effects through the control of DNA transcription and protein
synthesis. Like its naturally secreted counterpart, levothyroxine is a chiral compound
in the L-form.
|
Liothyronine Sodium
https://en.wikipedia.org/wiki/Liothyronine

Liothyronine is
a manufactured form of the thyroid hormone triiodothyronine (T3). It
is most commonly used to treat hypothyroidism and myxedema
coma.Liothyronine is the most potent form of thyroid hormone. As a salt of triiodothyronine (T3),
it is chemically similar and pharmacologically equivalent to T3. As such, it acts on the body to increase the basal metabolic rate, affect protein synthesis and increase the body's sensitivity to catecholamines (such as adrenaline) by permissiveness.
|
| Vitamin D Analogs |
Calcitriol
https://en.wikipedia.org/wiki/Calcitriol

Calcitriol is
the active form of vitamin D,
normally made in the kidney A
manufactured form is used to treat kidney disease with low
blood calcium, hyperparathyroidism due
to kidney disease, low blood calcium due to hypoparathyroidism, osteoporosis, osteomalacia,
and familial hypophosphatemia.
Many of the effects of calcitriol are mediated by its interaction with the calcitriol receptor, also called the vitamin D receptor or VDR. For instance, the unbound inactive form of the calcitriol receptor in intestinal epithelial cells resides in the cytoplasm. When calcitriol binds to the receptor, the ligand-receptor complex translocates to the cell
nucleus, where it acts as a transcription factor promoting the expression of a gene encoding a calcium binding protein. The levels of the calcium binding protein increase enabling the cells to actively transport more calcium (Ca2+
) from the intestine across the intestinal mucosa into the blood.
The maintenance of electroneutrality requires that the transport of Ca2+
ions catalyzed by the intestinal epithelial cells be accompanied by counterions, primarily inorganic phosphate. Thus calcitriol also stimulates the intestinal absorption of phosphate.
|
| |
|
| Immunology |
| Immuno-Suppressants |
Everolimus
https://en.wikipedia.org/wiki/Everolimus

Everolimus is
a medication used
as an immunosuppressant to
prevent rejection of organ
transplants and in the
treatment of renal cell cancer and other tumours.
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex. This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2
positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types. Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.
|
Mycophenolic Mofetil / Sodium
https://en.wikipedia.org/wiki/Mycophenolic_acid
https://en.wikipedia.org/wiki/Morpholino

Mycophenolic acid (MPA)
is an immunosuppressant medication used
to prevent rejection following organ
transplantation and to
treat Crohn's disease.[4] Specifically
it is used following kidney, heart,
and liver transplantation.Purines
(including the nucleotides guanosine and adenosine)
can either be synthesized de novo using ribose
5-phosphate or they can be salvaged from free nucleotides. Mycophenolic acid is potent, reversible, non-competitive inhibitor of inosine-5′-monophosphate
dehydrogenase (IMPDH), an enzyme essential to the de novo synthesis of guanosine-5'-monophosphate (GMP)
from inosine-5'-monophosphate (IMP). IMPDH inhibition particularly affects lymphocytes since
they rely almost exclusively on de novo purine synthesis. In contrast, many other cell types use both pathways, and some cells, such as terminally differentiated neurons, depend completely on purine nucleotide salvage. Thus, use of mycophenolic acid leads to a relatively selective inhibition of DNA
replication in T cells and B
cells..
|
Pimecrolimus
https://en.wikipedia.org/wiki/Pimecrolimus

Pimecrolimus is
an immunomodulating agent of
the calcineurin inhibitor class
used in the treatment of atopic dermatitis (eczema). .
Pimecrolimus is an ascomycin macrolactam derivative. It has been shown in vitro that pimecrolimus binds to FKBP1A and also inhibits calcineurin Thus pimecrolimus inhibits T-cell activation by inhibiting the synthesis and release of cytokines from T-cells. Pimecrolimus also prevents the release of inflammatory cytokines and mediators from mast
cells.
Pimecrolimus, like tacrolimus, belongs to the ascomycin class of macrolactam immunosuppressives, acting by the inhibition of T-cell activation by the calcineurin pathway and inhibition of the release of numerous inflammatory cytokines, thereby preventing the cascade of immune and inflammatory signals. Pimecrolimus has a similar mode of action to that of tacrolimus but is more selective, with no effect on dendritic (Langerhans) cells. It has lower permeation through the skin than topical steroids or topical tacrolimus although
they have not been compared with each other for their permeation ability through mucosa. In addition, in contrast with topical steroids, pimecrolimus does not produce skin atrophy.
|
Thalidomide
https://en.wikipedia.org/wiki/Thalidomide

Thalidomide,
sold under the brand name Thalomid among
others, is a medication used to treat a number of cancers including multiple myeloma, graft-versus-host
disease, and a number of skin conditions including complications of leprosy. While
it has been used in a number of HIV associated
conditions, such use is associated with increased levels of the virus.The precise mechanism of action for thalidomide is not known, although efforts to identify thalidomide's teratogenic action generated 2,000 research papers and the proposal of 15 or 16 plausible mechanisms by the year 2000. As of 2015, the main theories were inhibition of the process of angiogenesis,
its inhibition of cereblon, a ubiquitin
ligase, and its ability to generate reactive oxygen species which in turn kills cells.
|
| Multiple Sclerosis |
Teriflunomide
https://en.wikipedia.org/wiki/Teriflunomide

Teriflunomide,
sold under the brand name Aubagio, is the active
metabolite of leflunomide.
Teriflunomide is an immunomodulatory drug inhibiting pyrimidine de novo synthesis by blocking the enzyme dihydroorotate dehydrogenase. It is uncertain whether this explains its effect on MS lesions.
Teriflunomide inhibits rapidly dividing cells, including activated T cells, which are thought to drive the disease process in MS. Teriflunomide may decrease the risk of infections compared to chemotherapy-like drugs because of its more-limited effects on the immune system.
|
| |
|
| Metabolism |
| Anti- Obesity |
Cetilistat
https://en.wikipedia.org/wiki/Cetilistat

Cetilistat is
a drug designed to treat obesity.
It acts in the same way as the older drug orlistat (Xenical)
by inhibiting pancreatic lipase,
an enzyme that
breaks down triglycerides in
the intestine.
Without this enzyme, triglycerides from the diet are prevented from being hydrolyzed into
absorbable free fatty acids and
are excreted undigested.
|
Ezetimibe
https://en.wikipedia.org/wiki/Ezetimibe

Ezetimibe is
a medication used to treat high blood cholesterol and
certain other lipid abnormalities. Generally
it is used together with dietary changes and a statin.Ezetimibe
inhibits the absorption of cholesterol from the small intestine and
decreases the amount of cholesterol normally available to liver cells. This leads them to absorb more cholesterol from circulation and thus causes lowering levels of circulating cholesterol. It blocks the critical mediator of cholesterol absorption, the Niemann-Pick C1-like 1 (NPC1L1) protein on the gastrointestinal
tract epithelial cells, as well as in hepatocytes;
it blocks aminopeptidase N and interrupts a caveolin
1–annexin A2 complex
involved in trafficking cholesterol.
|
| Drugs used in Gout |
Colchicine
https://en.wikipedia.org/wiki/Colchicine
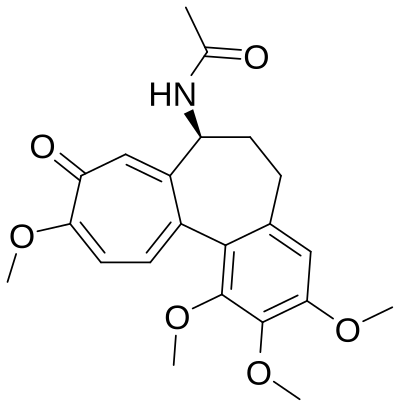
Colchicine is
a medication used to treat gout and Behçet's
disease. In
gout, it is less preferred to NSAIDs or steroids. Other
uses for colchicine include the prevention of pericarditis and familial
Mediterranean fever.In gout, inflammation in joints results
from the precipitation of circulating uric acid, exceeding its solubility in
blood and depositing as crystals of monosodium urate in and around synovial fluid and soft
tissues of joints.These crystal deposits cause inflammatory arthritis, which is initiated and sustained by mechanisms involving various proinflammatory mediators, such as cytokines. Colchicine
accumulates in white blood cells and affects them in a variety of ways: decreasing motility, mobilization (especially chemotaxis) and adhesion.
|
Febuxostat
https://en.wikipedia.org/wiki/Febuxostat

Febuxostat,
sold under the brand names Uloric and Adenuric among
others, is a medication used long-term to treat gout due
to high uric acid levels.Febuxostat
is a non-purine-selective inhibitor of xanthine oxidase. It
works by non-competitively blocking the molybdenum
pterin center, which is the active site of xanthine oxidase. Xanthine oxidase is needed to oxidize successively hypoxanthine and xanthine to
uric acid. Thus, febuxostat inhibits xanthine oxidase, thereby reducing production of uric acid. Febuxostat inhibits both the oxidized and the reduced forms of xanthine oxidase by virtue of its tight binding to the molybdenum pterin site.
|
Probenecid
https://en.wikipedia.org/wiki/Probenecid

Probenecid, also sold under the brand name Probalan, is a medication that increases uric acid excretion in the urine. It is primarily used in treating gout and hyperuricemia.
Probenecid was developed as an alternative to caronamide to competitively inhibit renal excretion of some drugs, thereby increasing their plasma concentration and prolonging their effects.Probenecid probably has several pharmacological targets, including blocking pannexins. Probenecid
is also useful in the treatment of gout where the mechanism of action is believed to be focused on the kidney. Probenecid interferes with the kidneys' organic anion transporter (OAT),
which reclaims uric acid from the urine and returns it to the plasma. If
probenecid (an organic acid) is present, the OAT binds preferentially to it (instead of to uric acid), preventing reabsorption of the uric acid. Hence, the urine retains more uric acid, lowering uric acid concentration in the plasma. This is a good example of a medical usage for competition between substrates transported across cell membranes. This same effect, however, alters excretion of acidic drugs by the kidney, leading to the many drug interactions noted above.
|
| Hepatitis C (HCV) |
Daclatasvir Dihydrochloride
https://en.wikipedia.org/wiki/Daclatasvir

Daclatasvir,
sold under the trade name Daklinza, is a medication used
in combination with other medications to treat hepatitis C (HCV). The
other medications used in combination include sofosbuvir, ribavirin,
and interferon,
vary depending on the virus type and whether the person has cirrhosis.Daclatasvir
stops HCV viral RNA replication and protein translation by directly inhibiting HCV protein NS5A. NS5A is critical for HCV viral transcription and translation, and as of 2014 it appeared that resistance can arise to daclatasvir fairly swiftly.
|
Ledipasvir
https://en.wikipedia.org/wiki/Ledipasvir

Ledipasvir is
a drug for the treatment of hepatitis C that
was developed by Gilead Sciences.
Ledipasvir inhibits an important viral phosphoprotein, NS5A, which is involved in viral replication, assembly, and secretion.
Sofosbuvir, on the other hand, is metabolized to a uridine triphosphate mimic, which acts as a RNA chain terminator when incorporated into RNA by NS5B polymerase.
|
Sofosbuvir
https://en.wikipedia.org/wiki/Sofosbuvir

Sofosbuvir,
sold under the brand name Sovaldi among
others, is a medication used to treat hepatitis C. It
is only recommended with some combination of ribavirin, peginterferon-alfa, simeprevir, ledipasvir, daclatasvir,
or velpatasvir. Cure
rates are 30 to 97% depending on the type of hepatitis C virus involved.
Sofosbuvir inhibits the hepatitis C NS5B protein. Sofosbuvir appears to have a high barrier to the development of resistance.
Sofosbuvir is a prodrug of the Protide type, whereby the active phosphorylated nucleotide is granted cell permeability and oral bioavailability. It is metabolized to the active antiviral agent GS-461203 (2'-deoxy-2'-α-fluoro-β-C-methyluridine-5'-triphosphate). GS-461203 serves as a defective substrate for the NS5B protein, which is the viral RNA polymerase, thus acts as an inhibitor of viral RNA synthesis. Although sofosbuvir has a 3' hydroxyl group to act as a nucleophile for an incoming NTP, a similar nucleotide analogue, 2'-deoxy-2'-α-fluoro-β-C-methylcytidine, is proposed to act as a chain terminator because the 2' methyl
group of the nucleotide analogue causes a steric clash with an incoming NTP. Sofosbuvir would act in a similar way.
|
Telaprevir
https://en.wikipedia.org/wiki/Telaprevir

Telaprevir (VX-950),
marketed under the brand names Incivek and Incivo,
is a pharmaceutical drug for the treatment of hepatitis C co-developed
by Vertex Pharmaceuticals and Johnson
& Johnson. It is a member of a class of antiviral
drugs known as protease
inhibitors. Specifically,
telaprevir inhibits the hepatitis C viral enzyme NS3/4A serine protease. Telaprevir
is only indicated for use against hepatitis C genotype 1
viral infections and has not been proven to have an effect on or being safe when used for other genotypes of the virus. The standard therapy of pegylated interferon and ribavirin is
less effective than telaprevir in those with genotype 1.
|
| Wilson’s Disease |
Trientine HCl
https://en.wikipedia.org/wiki/Triethylenetetramine

Triethylenetetramine (TETA and trien),
also called trientine (INN),
is an organic compound with
the formula [CH2NHCH2CH2NH2]2.
This oily liquid is colorless but, like many amines,
assumes a yellowish color due to impurities resulting from air-oxidation. It is soluble in polar solvents. The branched isomer tris(2-aminoethyl)amine and piperazine derivatives
may also be present in commercial samples of TETA.The hydrochloride salt of TETA, referred to as trientine hydrochloride, is a chelating
agent that is used to bind and remove copper in the body to treat Wilson's
disease, particularly in those who are intolerant to penicillamine.
|
| |
|
| Musculo - skeletal disorder |
| Arthritis |
Celecoxib
https://en.wikipedia.org/wiki/Celecoxib

Celecoxib,
sold under the brand name Celebrex among
others, is a COX-2 inhibitor and nonsteroidal
anti-inflammatory drug (NSAID). It
is used to treat the pain and inflammation in osteoarthritis, acute
pain in adults, rheumatoid
arthritis, ankylosing
spondylitis, painful
menstruation, and juvenile
rheumatoid arthritis. It
may also be used to decrease the risk of colorectal adenomas in
people with familial adenomatous polyposis.A
highly selective reversible inhibitor of the COX-2 isoform
of cyclooxygenase, celecoxib inhibits the transformation of arachidonic acid to prostaglandin precursors. Therefore, it has analgesic and anti-inflammatory properties.
For its use in reducing colon polyps, celecoxib affects genes and pathways involved in inflammation and malignant transformation in tumors, but not normal tissues.
Celecoxib binds to Cadherin-11 (which may explain the reduction in cancer progression).
|
Diacerein
https://en.wikipedia.org/wiki/Diacerein
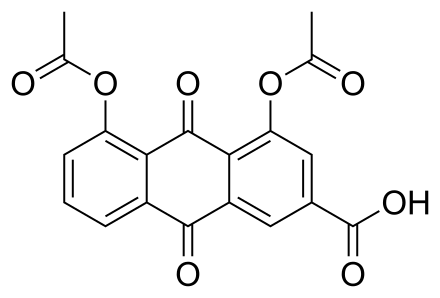
Diacerein (INN),
also known as diacetylrhein, is a slow-acting medicine of
the class anthraquinone used
to treat joint diseases such as osteoarthritis (swelling and pain in the joints). It
works by inhibiting interleukin-1 beta.Diacerein
works by blocking the actions of interleukin-1 beta,
a protein involved in the inflammation and destruction of cartilage that play a role in the development of symptoms of degenerative joint diseases such as osteoarthritis.
Due to its specific mode of action, which does not involve the inhibition of prostaglandin synthesis, diacerein has been shown to have anti-osteoarthritis and cartilage stimulating properties in vitro and animal models.
|
Risedronate
https://en.wikipedia.org/wiki/Risedronic_acid

Risedronic acid,
often used as its sodium salt risedronate sodium, is a bisphosphonate used
to strengthen bone, treat
or prevent osteoporosis,
and treat Paget's disease of bone. |
Sulfasalazine
https://en.wikipedia.org/wiki/Sulfasalazine

Sulfasalazine (SSZ),
sold under the trade name Azulfidine among
others, is a medication used to treat rheumatoid arthritis, ulcerative
colitis, and Crohn's
disease. It
is considered by some to be a first line treatment in rheumatoid arthritis.Sulfasalazine is in the disease-modifying antirheumatic drugs (DMARDs) family of medications. It is unclear exactly how it works. One proposed mechanism is the inhibition of prostaglandins,
resulting in local anti-inflammatory effects in the colon. The medication is broken down by intestinal
bacteria into sulfapyridine and 5-aminosalicylic
acid. That which is absorbed is excreted by the kidneys and in the bile.
|
Zinc Bisglycinate
https://en.wikipedia.org/wiki/Magnesium_glycinate
https://pubchem.ncbi.nlm.nih.gov/compound/151910#section=Uses
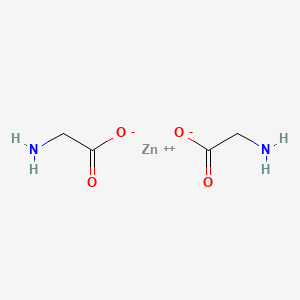
Magnesium glycinate, also known as magnesium
diglycinate or magnesium
bisglycinate, is the magnesium salt of glycine (one
magnesium and two glycine molecules), and is sold as a dietary supplement.Drug, dietary_supplement, Pharmaceutical related Food_additive, Includes spices, extracts, colorings, flavors, etc added to food for human consumption Personal care, including cosmetics, shampoos, perfumes, soaps, lotions, toothpastes, etc.
|
| Muscular Dystrophy, Duchenne |
Oxatomide Anhydrouse / Monohydrate
https://en.wikipedia.org/wiki/Oxatomide
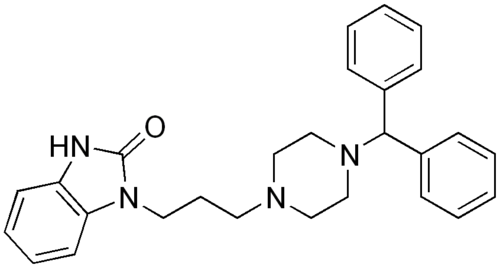
Oxatomide,
sold under the brand name Tinset among
others, is a first-generation antihistamine of
the diphenylmethylpiperazine family
which is marketed in Europe, Japan,
and a number of other countries. It
was discovered at Janssen Pharmaceutica in
1975. Oxatomide lacks
any anticholinergic effects. In
addition to its H1 receptor antagonism,
it also possesses antiserotonergic activity
similarly to hydroxyzine.
|
| Muscle Relaxants |
Flavoxate HCl
https://en.wikipedia.org/wiki/Flavoxate

Flavoxate is
an anticholinergic with antimuscarinic effects.
Its muscle relaxant properties
may be due to a direct action on the smooth muscle rather than by antagonizing muscarinic receptors. |
Meprobamate
https://en.wikipedia.org/wiki/Meprobamate

Meprobamate—marketed
as Miltown by Wallace
Laboratories and Equanil by Wyeth,
among others—is a carbamate derivative
used as an anxiolytic drug. Meprobamate's
mechanism of action is not completely known. It has been shown in animal studies to have effects at multiple sites in the central nervous system, including the thalamus and limbic
system. Meprobamate binds to GABAA receptors which
interrupts neuronal communication in the reticular formation and spinal
cord, causing sedation and altered perception of pain. Meprobamate has the ability to activate currents even in the absence of GABA. This relatively unique property makes meprobamate exceptionally dangerous when used in combination with other GABA-mediated drugs (including alcohol). It is also a potent adenosine reuptake inhibitor. |
Methocarbamol
https://en.wikipedia.org/wiki/Methocarbamol

Methocarbamol, sold under the brand name Robaxin among
others, is a medication used for short-term musculoskeletal pain.The
mechanism of action of methocarbamol has not currently been established. Its effect is thought to be localized to the central nervous system rather than a direct effect on skeletal muscles. It has no effect on the motor end plate or the peripheral nerve fiber. The efficacy of the medication is likely related to its sedative effect. Alternatively, methocarbamol may act via inhibition of acetylcholinesterase, similarly to carbamate..
|
Thiocolchicoside
https://en.wikipedia.org/wiki/Thiocolchicoside

Thiocolchicoside (Muscoril, Myoril, Neoflax)
is a muscle relaxant with anti-inflammatory and analgesic effects. It
acts as a competitive GABAA receptor antagonist and
also glycine receptor antagonist with
similar potency and nicotinic
acetylcholine receptors to
a much lesser extent. It
has powerful convulsant activity
and should not be used in seizure-prone
individuals. |
Tofisopam
https://en.wikipedia.org/wiki/Tofisopam

Tofisopam (Emandaxin, Grandaxin, Sériel)
is an anxiolytic that
is marketed in several European countries. Chemically,
it is a 2,3-benzodiazepine. Unlike other anxiolytic benzodiazepines (which
are generally 1,4- or 1,5-substituted) however, tofisopam does not have anticonvulsant, sedative, skeletal
muscle relaxant, motor
skill-impairing or amnestic properties.
While it may not be an anticonvulsant in and of itself, it has been shown to enhance the anticonvulsant action of classical 1,4-benzodiazepines (such as diazepam) and muscimol,
but not sodium valproate, carbamazepine, phenobarbital,
or phenytoin.Tofisopam is
indicated for the treatment of anxiety and alcohol
withdrawal, and is prescribed in a dosage of 50–300 mg per day divided into three doses.
|
| Muscle Spasms |
Baclofen
https://en.wikipedia.org/wiki/Baclofen

Baclofen,
sold under the brand name Lioresal among
others, is a medication used
to treat muscle spasticity such
as from a spinal cord injury or multiple
sclerosis. It
may also be used for hiccups and muscle
spasms near the end of
life.Chemically, baclofen is a derivative of the neurotransmitter γ-aminobutyric
acid (GABA). It is believed to work by activating (or agonizing) GABA receptors, specifically the GABAB receptors. Its
beneficial effects in spasticity result from its actions in the brain and spinal
cord. |
| Nonopioid Analgesics |
Carbamazepine
https://en.wikipedia.org/wiki/Carbamazepine
Carbamazepine (CBZ),
sold under the trade name Tegretol among
others, is an anticonvulsant medication used
primarily in the treatment of epilepsy and neuropathic
pain. It
is used in schizophrenia along
with other medications and as a second-line agent in bipolar disorder. Carbamazepine
appears to work as well as phenytoin and valproate for
focal and generalized seizures It
is not effective for absence or myoclonic
seizures.Carbamazepine is a sodium
channel blocker. It binds preferentially to voltage-gated sodium channels in their inactive conformation, which prevents repetitive and sustained firing of an action potential. Carbamazepine has effects on serotonin systems but the relevance to its antiseizure effects is uncertain. There is evidence that it is a serotonin releasing agent and possibly even a serotonin
reuptake inhibitor.
|
Etodolac
https://en.wikipedia.org/wiki/Etodolac

Etodolac is
a nonsteroidal anti-inflammatory drug (NSAID).NSAIDs
are used for the management of mild to moderate pain, fever, and inflammation.
They work by reducing the levels of prostaglandins, which are chemicals that are responsible for pain and the fever and tenderness that occur with inflammation. Etodolac blocks the cyclooxygenase (abbrev.
COX) enzymes which form prostanoids, resulting in lower concentrations of prostaglandins.
As a consequence, inflammation, pain and fever are reduced.
|
Glucosamine Sulphate
https://en.wikipedia.org/wiki/Glucosamine

Glucosamine (C6H13NO5) is an amino sugar and a prominent precursor in the biochemical synthesis of glycosylated proteins and lipids. Glucosamine is part of the
structure of the polysaccharides, chitosan, and chitin. Glucosamine is one of the most abundant monosaccharides. Produced commercially by the hydrolysis of shellfish exoskeletons or,
less commonly, by fermentation of a grain such as corn or wheat, glucosamine has many names depending on country.
Although a common dietary supplement, there is little evidence that it is effective for relief of arthritis or pain, and is not an approved prescription drug..
|
Indomethacin
https://en.wikipedia.org/wiki/Indometacin

Indometacin,
also known as indomethacin, is a nonsteroidal
anti-inflammatory drug (NSAID)
commonly used as a prescription medication to
reduce fever, pain, stiffness,
and swelling from inflammation.
It works by inhibiting the production of prostaglandins, endogenous signaling
molecules known to cause
these symptoms. It does this by inhibiting cyclooxygenase,
an enzyme that catalyzes the
production of prostaglandins.Indometacin, a non-steroidal anti-inflammatory drug (NSAID), has similar mode of action when compared to other drugs in this group. Its is a nonselective inhibitor of cyclooxygenase (COX) 1 and 2, the enzymes that participate in prostaglandin synthesis from arachidonic
acid. Prostaglandins are hormone-like molecules normally found in the body, where they have a wide variety of effects, some of which lead to pain, fever, and inflammation. By inhibiting the synthesis of prostaglandins, indometacin can reduce pain, fever, and inflammation. Indometacin mechanism of action, along with several other NSAIDs that inhibit COX, was described in 1971. |
Ketorolac Tromethamine
https://en.wikipedia.org/wiki/Ketorolac
https://en.wikipedia.org/wiki/Tris .
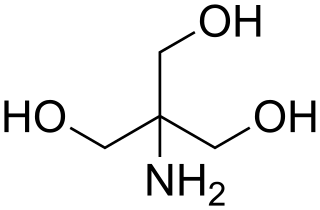
Ketorolac,
sold under the brand name Toradol among
others, is a nonsteroidal anti-inflammatory drug (NSAID)
used to treat pain. Specifically
it is recommended for moderate to severe pain.
Tris (usually known as THAM in this context) is used as alternative to sodium bicarbonate in the
treatment of metabolic acidosis. |
Leflunomide
https://en.wikipedia.org/wiki/Leflunomide

Leflunomide,
sold under the brand name Arava among
others, is an immunosuppressive disease-modifying antirheumatic drug (DMARD), used
in active moderate-to-severe rheumatoid arthritis and psoriatic
arthritis. It is a pyrimidine
synthesis inhibitor that
works by inhibiting dihydroorotate dehydrogenase.
Leflunomide is an immunomodulatory drug that achieves its effects by inhibiting the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH), which plays a key role in the de novo synthesis of uridine monophosphate (rUMP), which is required for the synthesis of DNA and RNA. Hence, leflunomide inhibits the reproduction of rapidly dividing cells, especially lymphocytes.
The inhibition of human DHODH by teriflunomide, the active metabolite of leflunomide, occurs at levels (approximately 600 nM) that are achieved during treatment of rheumatoid arthritis (RA).
|
Meloxicam
https://en.wikipedia.org/wiki/Meloxicam

Meloxicam,
sold under the brand name Mobic among
others, is a nonsteroidal anti-inflammatory drug (NSAID)
used to treat pain and inflammation in rheumatic diseases and osteoarthritis.Meloxicam
blocks cyclooxygenase (COX),
the enzyme responsible for converting arachidonic
acid into prostaglandin
H2—the first step in the synthesis of prostaglandins, which are mediators of inflammation. Meloxicam has been shown, especially at its low
therapeutic doses, selectively to inhibit COX-2 over COX-1..
|
Nimesulide
https://en.wikipedia.org/wiki/Nimesulide

Nimesulide is
a nonsteroidal anti-inflammatory drug (NSAID)
with pain medication and fever
reducing properties. Its
approved indications are the treatment of
acute pain, the symptomatic treatment of osteoarthritis,
and primary dysmenorrhoea in adolescents and
adults above 12 years old.
|
Oxcarbazepine
https://en.wikipedia.org/wiki/Oxcarbazepine

Oxcarbazepine,
sold under the brand name Trileptal among
others, is a medication used to treat epilepsy and bipolar
disorder. For
epilepsy it is used for both focal seizures and generalized
seizures. It
has been used both alone and as add-on therapy in people with bipolar who have had no success with other treatments. It
is taken by mouth.Oxcarbazepine is a prodrug, which is largely metabolised to
its pharmacologically active 10-monohydroxy derivative licarbazepine (sometimes abbreviated MHD). Oxcarbazepine and MHD exert their action by blocking voltage-sensitive sodium channels, thus leading to the stabilization of hyper-excited neural membranes, suppression of repetitive neuronal firing and diminishment propagation of synaptic impulses. Furthermore, anticonvulsant effects of these compounds could be attributed to enhanced potassium conductance and modulation of high-voltage activated calcium channels.
|
Paracetamol / DC Grade
https://en.wikipedia.org/wiki/Paracetamol

Paracetamol,
also known as acetaminophen, is a medication used to treat pain and fever. It
is typically used for mild to moderate pain relief. Evidence
is mixed for its use to relieve fever in children. It
is often sold in combination with other medications, such as in many cold medications. Paracetamol
is also used for severe pain, such as cancer pain and
pain after surgery, in combination with opioid pain medication.Despite
its common use, the mechanism of action of paracetamol is not completely understood. Unlike NSAIDs such as aspirin, paracetamol does not appear to inhibit the function of any cyclooxygenase (COX)
enzyme outside the central nervous system, and this appears to be the reason why it is not useful as an anti-inflammatory. It
does appear to selectively inhibit COX activities in the brain, which may contribute to its ability to treat fever and pain. This activity does not appear to be direct inhibition by blocking an active site, but rather by reducing COX, which must be oxidized in order to function. |
Piroxicam
https://en.wikipedia.org/wiki/Piroxicam

Piroxicam is
a nonsteroidal anti-inflammatory drug (NSAID)
of the oxicam class
used to relieve the symptoms of painful inflammatory conditions like arthritis. Piroxicam
works by preventing the production of endogenous prostaglandins which
are involved in the mediation of pain, stiffness, tenderness and swelling.Piroxicam is an NSAID and, as such, is a non-selective COX inhibitor possessing both analgesic and antipyretic properties. |
| Opioid Analgesics |
Tramadol Hydrochloride
https://en.wikipedia.org/wiki/Tramadol
Tramadol,
sold under the brand name Ultram among
others, is an opioid pain
medication used to treat
moderate to moderately severe pain.Tramadol
induces analgesic effects through a variety of different targets on the noradrenergic system, serotoninergic
system and opioid receptors system. Tramadol exists as a racemic
mixture, the positive enantiomer inhibits serotonin reuptake while the negative enantiomer inhibits noradrenaline re-uptake, by binding to and blocking the transporters. Tramadol has also been shown to act as a serotonin releasing agent. Both enantiomers of tramadol are agonists of the μ-opioid
receptor and its M1 metabolite, O-demethylate, is also a μ-opioid receptor agonist but is 6 times more potent than tramadol itself. All these effects work synergistically to induce analgesia. |
| Osteoporosis |
Alendronate Sodium
https://en.wikipedia.org/wiki/Alendronic_acid

Alendronic acid, sold under the brand name Fosamax among
others, is a bisphosphonate medication used
to treat osteoporosis and Paget's
disease of bone. It
is taken by mouth. Use
is often recommended together with vitamin D, calcium
supplementation, and lifestyle changes.
Alendronate inhibits osteoclast-mediated bone-resorption. Like all bisphosphonates, it is chemically related to inorganic pyrophosphate, the endogenous regulator of bone turnover. But while pyrophosphate inhibits both osteoclastic bone resorption and the mineralization of the bone newly formed by osteoblasts,
alendronate specifically inhibits bone resorption without any effect on mineralization at pharmacologically achievable doses. Its inhibition of bone-resorption is dose-dependent and approximately 1,000 times stronger than the equimolar effect of the first bisphosphonate drug, etidronate. Under therapy, normal bone tissue develops, and alendronate is deposited in the bone-matrix in a pharmacologically inactive form. For optimal action, enough calcium and vitamin D are needed in the body in order to promote normal bone development. Hypocalcemia should, therefore, be corrected before starting therapy.
Etidronate has the same disadvantage as pyrophosphate in inhibiting mineralization, but all of the potent N-containing bisphosphonates, including alendronate, risedronate, ibandronate, and zoledronate, do not.
|
Ambroxol HCl/Base
https://en.wikipedia.org/wiki/Ambroxol

Ambroxol is
a drug that breaks up phlegm, used in the treatment of respiratory
diseases associated with
viscid or excessive mucus. Recently, a hypothesis suggested that it may have a potential role in treatment of Paget's
disease of bone, Parkinsonism,
and other common diseases of aging-associated diseases involving
dysfunction of autophagy. Ambroxol
is often administered as an active ingredient in cough syrup.
The substance acts on mucus membranes, restoring the physiological clearance mechanisms of the respiratory tract (which play an important role in the body’s natural defence mechanisms) through several mechanisms, including breaking up phlegm, stimulating mucus production, and stimulating synthesis and release of surfactant by
type II pneumocytes. Surfactant acts as an anti-glue factor by reducing the adhesion of mucus to the bronchial wall, in improving its transport and in providing protection against infection and irritating agents.
Ambroxol is a potent inhibitor of the neuronal Na+ channels. This property led to the development of a lozenge containing 20 mg of ambroxol. Many state-of-the-art clinical studies have demonstrated the efficacy of ambroxol in relieving pain in acute sore throat, with a rapid onset of action, with its effect lasting at least three hours. Ambroxol is also anti-inflammatory,
reducing redness in a sore throat.
Ambroxol has recently been shown to increase activity of the lysosomal enzyme glucocerebrosidase. Because of this it may be a useful therapeutic agent for both Gaucher disease and Parkinson's disease.
It was also recently shown that ambroxol triggers exocytosis of lysosomes by releasing calcium from acidic cellular calcium stores. This occurs by diffusion of ambroxol into lysosomes and lysosomal pH neutralization. This mechanism is most likely responsible for the mucolytic effects of the drug, but may also explain the reported activity in Gaucher and Parkinson's disease.
Both ambroxol and its parent drug bromhexine have been shown to induce autophagy in several cell types, and ambroxol was shown to potentiate rifampicin therapy in a model of tuberculosis through host directed effects. Ambroxol also enhances lung levels of a wide range of antibiotics. Ambroxol also had activity against SARS CoV2 in Vero E6 cells, although the mechanism of action is not yet known.
|
| |
|
| Oncology |
| Antimucositis |
Rutoside
https://en.wikipedia.org/wiki/Rutin

Rutin,
also called rutoside, quercetin-3-O-rutinoside and sophorin,
is the glycoside combining
the flavonol quercetin and
the disaccharide rutinose (α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranose).
It is a citrus flavonoid found
in a wide variety of plants including citrus.Rutin is a citrus flavonoid glycoside found
in many plants including buckwheat, the leaves and petioles of Rheum species,
and asparagus. Tartary
buckwheat seeds have been found to contain more rutin (about 0.8–1.7% dry weight) than common buckwheat seeds (0.01% dry weight). Rutin is one of the primary flavonols found in 'clingstone' peaches. It is also found in green
tea infusions. |
| Antineoplastic Agents |
Irinotecan HCl
https://en.wikipedia.org/wiki/Irinotecan

Irinotecan,
sold under the brand name Camptosar among
others, is a medication used to treat colon cancer,
and small cell lung cancer. For
colon cancer it is used either alone or with fluorouracil.For
small cell lung cancer it is used with cisplatin.
Camptothecin, one of the four major structural classifications of plant-derived anti-cancerous compounds, is a cytotoxic alkaloid which consists of a pentacyclic ring structure containing a pyrrole (3, 4 β) quinoline moiety, an S-configured lactone form, and a carboxylate form. Irinotecan is activated by hydrolysis to SN-38, an inhibitor of topoisomerase I. This is then inactivated by glucuronidation by uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1).
The inhibition of topoisomerase I by the active metabolite SN-38 eventually leads to inhibition of both DNA replication and transcription.
The molecular action of irinotecan occurs by trapping a subset of topoisomerase-1-DNA cleavage complexes, those with a guanine +1 in the DNA sequence. One irinotecan molecule stacks against the base pairs flanking the topoisomerase-induced cleavage site and poisons (inactivates) the topoisomerase 1 enzyme.
|
Mitomycin
https://en.wikipedia.org/wiki/Mitomycins

The mitomycins are
a family of aziridine-containing natural
products isolated from Streptomyces
caespitosus or Streptomyces
lavendulae. They
include mitomycin A, mitomycin
B, and mitomycin C.
When the name mitomycin occurs alone, it usually refers to mitomycin C, its international nonproprietary name.
Mitomycin C is used as a medicine for treating various disorders associated with the growth and spread of cells.Mitomycin C has been shown to have activity against stationary phase persisters caused
by Borrelia burgdorferi, a factor in lyme
disease. Mitomycin C is used to treat symptoms of pancreatic and stomach
cancer, and is under clinical research for its potential to treat gastrointestinal strictures, wound
healing from glaucoma surgery, corneal excimer laser surgery and endoscopic dacryocystorhinostomy.
|
| Bladder,Kidney & other Urologic Cancers |
Sunitinib
https://en.wikipedia.org/wiki/Sunitinib

Sunitinib (marketed
as Sutent by Pfizer,
and previously known as SU11248) is an oral,
small-molecule, multi-targeted receptor tyrosine kinase (RTK)
inhibitor that was approved by the FDA for the treatment of renal cell carcinoma (RCC)
and imatinib-resistant gastrointestinal
stromal tumor (GIST) on
January 26, 2006. Sunitinib was the first cancer drug simultaneously approved for two different indications.
Sunitinib inhibits cellular signaling by targeting multiple receptor tyrosine kinases (RTKs).
These include all receptors for platelet-derived growth factor (PDGF-Rs) and vascular endothelial growth factor receptors (VEGFRs),
which play a role in both tumor angiogenesis and tumor cell proliferation. The simultaneous inhibition of these targets therefore reduces tumor vascularization and triggers cancer cell apoptosis and thus results in tumor shrinkage.
Sunitinib also inhibits CD117 (c-KIT), the receptor tyrosine kinase that (when improperly activated by mutation) drives the majority of gastrointestinal stromal cell tumors.
|
| Brain Cancer, Head and Neck Cancer, Lung Cancer, Ovarian Cancer & Neuroblastoma |
Carboplatin
https://en.wikipedia.org/wiki/Carboplatin

Carboplatin,
sold under the trade name Paraplatin among
others, is a chemotherapy medication
used to treat a number of forms of cancer. This
includes ovarian cancer, lung
cancer, head
and neck cancer, brain
cancer, and neuroblastoma.
Two theories exist to explain the molecular mechanism of action of carboplatin with DNA:
- Aquation, or the like-cisplatin hypothesis.
- Activation, or the unlike-cisplatin hypothesis.
The former is more accepted owing to the similarity of the leaving groups with its predecessor cisplatin, while the latter hypothesis envisages a biological activation mechanism to release the active Pt2+ species.
|
| Chemotherapeutic Agents |
Azacitidine
https://en.wikipedia.org/wiki/Azacitidine

Azacitidine,
sold under the brand name Vidaza among
others, is a chemical analog of cytidine,
a nucleoside in DNA and RNA.
Azacitidine and its deoxy derivative, decitabine (also
known as 5-aza-2′-deoxycytidine), are used in the treatment of myelodysplastic syndrome.
Azacitidine is a chemical analogue of the nucleoside cytosine, which is present in DNA and RNA. It is thought to have antineoplastic activity via two mechanisms – at low doses, by inhibiting of DNA methyltransferase, causing hypomethylation of DNA, and at high doses, by its direct cytotoxicity to abnormal hematopoietic cells in the bone marrow through its incorporation into DNA and RNA, resulting in cell death.
|
Cisplatin
https://en.wikipedia.org/wiki/Cisplatin

Cisplatin is
a chemotherapy medication used
to treat a number of cancers.Cisplatin
is in the platinum-based antineoplastic family
of medications. It works in part by binding to DNA and inhibiting its replication.Cisplatin interferes with DNA replication, which kills the fastest proliferating cells, which in theory are cancerous. Following administration, one chloride ion is slowly displaced by water to give the aquo
complex cis-[PtCl(NH3)2(H2O)]+,
in a process termed aquation. Dissociation of the chloride is favored inside the cell because the intracellular chloride concentration is only 3–20% of the approximately 100 mM chloride concentration in the extracellular fluid.
|
Dactinomycin
https://en.wikipedia.org/wiki/Dactinomycin

Dactinomycin,
also known as actinomycin D, is a chemotherapy
medication used to treat a
number of types of cancer.
Because actinomycin can bind DNA duplexes, it can also interfere with DNA replication, although other chemicals such as hydroxyurea are better suited for use in the laboratory as inhibitors of DNA synthesis.
In cell biology,
actinomycin D is shown to have the ability to inhibit transcription.
Actinomycin D does this by binding DNA at
the transcription initiation complex and preventing elongation of RNA chain
by RNA polymerase.Actinomycin
D and its fluorescent derivative, 7-aminoactinomycin D (7-AAD), are used as stains in microscopy and flow cytometry applications. The affinity of these stains/compounds for GC-rich regions of DNA strands makes them excellent markers for DNA. 7-AAD binds
to single stranded DNA; therefore it is a useful tool in determining apoptosis and distinguishing between dead cells and live ones.
|
Diminazene Aceturate
https://en.wikipedia.org/wiki/Diminazene

Diminazene (INN;
also known as diminazen) is an anti-infective medication for
animals that is sold under a variety of brand names. It is effective against certain protozoa such
as Babesia, Trypanosoma,
and Cytauxzoon.
The drug may also be effective against certain bacteria including Brucella and Streptococcus.
Chemically it is a di-amidine and it is formulated as its aceturate salt, diminazene aceturate.
The mechanism is not well understood; it probably inhibits DNA replication, but also has affinity to RNA.
|
Doxorubicin
https://en.wikipedia.org/wiki/Doxorubicin

Doxorubicin,
sold under the brand name Adriamycin among
others, is a chemotherapy medication used
to treat cancer.[3] This
includes breast cancer, bladder
cancer, Kaposi's
sarcoma, lymphoma,
and acute lymphocytic leukemia. It
is often used together with other chemotherapy agents.
Doxorubicin interacts with DNA by intercalation and inhibition of macromolecular biosynthesis. This inhibits the progression of topoisomerase II, an enzyme which relaxes supercoils in DNA for transcription. Doxorubicin
stabilizes the topoisomerase II complex after it has broken the DNA chain for replication, preventing the DNA double helix from being resealed and thereby stopping the process of replication.[4] It may also increase quinone type free radical production, hence contributing to its cytotoxicity.
The planar aromatic chromophore portion of the molecule intercalates between two base pairs of the DNA, while the six-membered daunosamine sugar sits in the minor groove and interacts with flanking base pairs immediately adjacent to the intercalation site, as evidenced by several crystal structures.
By intercalation, doxorubicin can also induce histone eviction from transcriptionally active chromatin. As a result, DNA damage response, epigenome and transcriptome are
deregulated in doxorubicin-exposed cells.
|
Epirubicin
https://en.wikipedia.org/wiki/Epirubicin

Epirubicin is an anthracycline drug used for chemotherapy. It can be used in combination with other medications to treat breast cancer in patients who have had surgery to remove the tumor. It is marketed by Pfizer under the trade name Ellence in the US and Pharmorubicin or Epirubicin
Ebewe elsewhere.
Similarly to other anthracyclines, epirubicin acts by intercalating DNA strands. Intercalation results in complex formation which inhibits DNA and RNA synthesis. It also triggers DNA cleavage by topoisomerase II, resulting in mechanisms that lead to cell death. Binding to cell membranes and plasma proteins may be involved in the compound's cytotoxic effects. Epirubicin also generates free
radicals that cause cell and DNA damage.
|
Gemcitabine HCl
https://en.wikipedia.org/wiki/Gemcitabine

Gemcitabine,
sold under the brand name Gemzar, among others,[1] is
a chemotherapy medication used
to treat a number of types of cancer.
Gemcitabine is hydrophilic and must be transported into cells via molecular transporters for nucleosides (the most common transporters for gemcitabine are SLC29A1 SLC28A1, and SLC28A3). After entering the cell, gemcitabine is first modified by attaching a phosphate to it, and so it becomes gemcitabine monophosphate (dFdCMP).This is the rate-determining
step that is catalyzed by the enzyme deoxycytidine kinase (DCK). Two more phosphates are added by other enzymes. After the attachment of the three phosphates gemcitabine is finally pharmacologically active as gemcitabine triphosphate (dFdCTP).
After being thrice phosphorylated, gemcitabine can masquerade as deoxycytidine triphosphate and is incorporated into new DNA strands being synthesized as the cell replicates.
|
Palbociclib
https://en.wikipedia.org/wiki/Palbociclib

Palbociclib,
sold under the brand name Ibrance among
others, is a medication developed by Pfizer for
the treatment of HR-positive and HER2-negative breast cancer.
It is a selective inhibitor of
the cyclin-dependent kinases CDK4 and CDK6. Palbociclib
was the first CDK4/6 inhibitor to be approved as a cancer therapy.
It is a selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6.
In the G1 phase of the cell cycle, mammalian cells must pass a checkpoint, known as the restriction point "R", in order to complete the cell cycle and divide. CDK4 and CDK6 complex with cyclin D drive the phosphorylation of
the retinoblastoma protein, Rb, which allows the cell to pass R and commit to division. Regulation of one or more proteins involved in this checkpoint is lost in many cancers. However, by inhibiting CDK4/6, palbociclib ensures that the cyclin D-CDK4/6 complex cannot aid in phosphorylating Rb. This prevents the cell from passing R and exiting G1, and in turn from proceeding through the cell cycle.
|
Risedronate
ttps://en.wikipedia.org/wiki/Risedronic_acid
Risedronic acid,
often used as its sodium salt risedronate sodium, is a bisphosphonate used
to strengthen bone, treat
or prevent osteoporosis,
and treat Paget's disease of bone.
|
| Colorectal Cancer |
Oxaliplatin
https://en.wikipedia.org/wiki/Oxaliplatin

Oxaliplatin,
sold under the brand name Eloxatin, is a cancer
medication used to treat colorectal
cancer. Often
it is used together with fluorouracil and folinic
acid (leucovorin) in
advanced cancer.According to in
vivo studies,
oxaliplatin fights carcinoma of the colon through
non-targeted cytotoxic effects. Like other platinum compounds, its cytotoxicity is thought to result from inhibition of DNA
synthesis in cells. In particular, oxaliplatin forms both inter- and intra-strand cross links in DNA, which prevent DNA replication and transcription, causing cell death.. |
| Hormone Therapy |
Letrozole
https://en.wikipedia.org/wiki/Letrozole

Letrozole,
sold under the brand name Femara among
others, is an aromatase inhibitor which
is used in the treatment of hormonally-responsive breast cancer after
surgery.Letrozole is an orally
active, nonsteroidal, selective aromatase
inhibitor and hence an antiestrogen.
It prevents aromatase from producing estrogens by competitive, reversible binding
to the heme of
its cytochrome P450 unit.
The action is specific, and letrozole does not reduce production of corticosteroids.
|
| Leukemias,Lymphomas & other Hematologic Cancers |
Romidepsin
https://en.wikipedia.org/wiki/Romidepsin

Romidepsin,
also known as Istodax, is an anticancer
agent used in cutaneous
T-cell lymphoma (CTCL) and
other peripheral T-cell lymphomas (PTCLs).
Romidepsin is a natural product obtained
from the bacterium Chromobacterium violaceum,
and works by blocking enzymes known as histone deacetylases,
thus inducing apoptosis. It
is sometimes referred to as depsipeptide,
after the class of molecules to which it belongs.Romidepsin acts as a prodrug with
the disulfide bond undergoing reduction within
the cell to release a zinc-binding thiol. The thiol binds to a zinc atom in the binding pocket of Zn-dependent histone
deacetylase to block its activity. Thus it is an HDAC inhibitor. Many HDAC
inhibitors are potential treatments for cancer through the ability to epigenetically restore normal expression of tumor suppressor genes, which may result in cell cycle arrest, differentiation, and apoptosis.
|
| Multiple Myeloma |
Bortezomib
https://en.wikipedia.org/wiki/Bortezomib
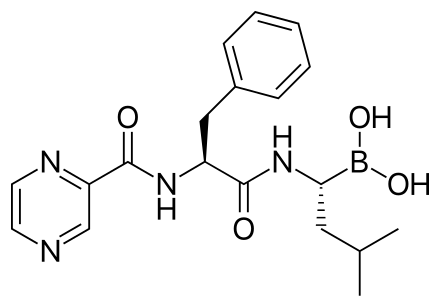
Bortezomib,
sold under the brand name Velcade among
others, is an anti-cancer medication used
to treat multiple myeloma and mantle
cell lymphoma. This
includes multiple myeloma in those who have and have not previously received treatment.The boron atom in bortezomib binds the catalytic site of the 26S
proteasome with high affinity and specificity. In normal cells, the proteasome regulates protein expression and function by degradation of ubiquitylated proteins, and also rids the cell of abnormal or misfolded proteins. Clinical and preclinical data support a role for the proteasome in maintaining the immortal phenotype of myeloma cells, and cell-culture and xenograft data support a similar function in solid tumor cancers. While multiple mechanisms are likely to be involved, proteasome inhibition may prevent degradation of pro-apoptotic factors, thereby triggering programmed cell death in neoplastic cells. Recently, it was found that bortezomib caused a rapid and dramatic change in the levels of intracellular peptides that are produced by the proteasome. Some intracellular peptides have been shown to be biologically active, and so the effect of bortezomib on the levels of intracellular peptides may contribute to the biological and/or side effects of the
drug.
|
Carfilzomib
https://en.wikipedia.org/wiki/Carfilzomib

Carfilzomib (marketed
under the trade name Kyprolis, developed by Onyx
Pharmaceuticals) is an anti-cancer drug acting as a selective proteasome
inhibitor. Chemically, it is a tetrapeptide epoxyketone and
an analog of epoxomicin.Carfilzomib
covalently irreversibly
binds to and inhibits the chymotrypsin-like
activity of the 20S proteasome,
an enzyme that degrades unwanted cellular proteins. Carfilzomib displays minimal interactions with non-proteasomal targets, thereby improving safety profiles over bortezomib. Inhibition
of proteasome-mediated proteolysis results in a build-up of polyubiquitinated proteins, which may cause cell cycle arrest, apoptosis,
and inhibition of tumor growth.
|
| Prostate Cancer |
Cabazitaxel
https://en.wikipedia.org/wiki/Cabazitaxel

Cabazitaxel (previously XRP-6258, trade name Jevtana) is a semi-synthetic derivative of a natural taxoid. It was developed by Sanofi-Aventis and was approved by the U.S. FDA for the treatment of hormone-refractory prostate
cancer on June 17, 2010. It is a microtubule inhibitor, and the fourth taxane to be approved as a cancer therapy.
Cabazitaxel in combination with prednisone is a treatment option for hormone-refractory prostate cancer following docetaxel-based treatment.
Taxanes enhance the microtubules stabilization and inhibit the cellular mitosis and division. Moreover,
taxanes prevent androgen receptor (AR) signaling by binding cellular microtubules and the microtubule-associated motor protein dynein, thus averting AR nuclear translocation.
|
Enzalutamide
https://en.wikipedia.org/wiki/Enzalutamide

Enzalutamide,
sold under the brand name Xtandi, is a nonsteroidal
antiandrogen (NSAA)
medication which is used in the treatment of prostate cancer. It
is indicated for use in conjunction with castration in
the treatment of metastatic castration-resistant
prostate cancer (mCRPC),[2] nonmetastatic
castration-resistant prostate cancer, and
metastatic castration-sensitive prostate cancer (mCSPC).Enzalutamide acts as a selective silent antagonist of
the androgen receptor (AR), the biological
target of androgens like testosterone and dihydrotestosterone (DHT).
Unlike the first-generation NSAA bicalutamide, enzalutamide does not promote translocation of
AR to the cell nucleus and in addition prevents binding of AR to deoxyribonucleic
acid (DNA) and AR to coactivator proteins. As
such, it has been described as an AR signaling inhibitor in addition to antagonist.The drug is described as a "second-generation" NSAA because it has greatly increased efficacy as an antiandrogen relative to so-called "first-generation" NSAAs like flutamide and bicalutamide. The drug has only 2-fold lower affinity for the AR than DHT, the endogenous ligand of the AR in the prostate gland.
|
| Renal Cell Carcinoma (RCC) |
Temsirolimus
https://en.wikipedia.org/wiki/Temsirolimus

Temsirolimus (codenamed CCI-779)
is an intravenous drug for the treatment of renal cell carcinoma (RCC)Temsirolimus
is a specific inhibitor of mTOR and interferes with the synthesis of proteins that regulate proliferation, growth, and survival of tumor cells. Though temsirolimus shows activity on its own, it is also known to be converted to sirolimus
(rapamycin) in vivo; therefore, its activity may be more attributed to its metabolite rather than the prodrug itself
(despite claims to the contrary by the manufacturer). Treatment with temsirolimus leads to cell cycle arrest in the G1
phase, and also inhibits tumor angiogenesis by reducing synthesis of VEGF.
|
| |
|
| Oropharyngeal |
| Nasal Decongestants |
Fluticasone Propionate / Furoate
https://en.wikipedia.org/wiki/Fluticasone_propionate
Fluticasone propionate,
sold under the brand names Flovent and Flonase among
others, is a steroid medication. When
inhaled it is used for the long term management of asthma and COPD. In
the nose it is used for hay fever and nasal
polyps. It
can also be used for mouth ulcers.Fluticasone
propionate is a highly selective agonist at
the glucocorticoid receptor with negligible activity at androgen, estrogen,
or mineralocorticoid receptors, thereby producing anti-inflammatory and vasoconstriction effects.
It has been shown to have a wide range of inhibitory effects on multiple cell types (e.g. mast cell, eosinophil, neutrophil, macrophages,
and lymphocytes) and mediators (e.g. histamine, eicosanoids, leukotrienes,
and cytokines) involved in inflammation.
Fluticasone propionate is stated to exert a topical effect on the lungs without significant systemic effects at usual doses, due to its low systemic bioavailability. |
Phenylephrine
https://en.wikipedia.org/wiki/Phenylephrine
Phenylephrine is
a medication primarily used as a decongestant,
to dilate the pupil,
to increase blood pressure,
and to relieve hemorrhoids.
Phenylephrine is a sympathomimetic drug, which means that it mimics the actions of epinephrine (commonly known as adrenaline) or norepinephrine. Phenylephrine selectively binds to alpha-1 receptors which cause blood vessels to constrict.[21]
Whereas pseudoephedrine causes both vasoconstriction and increase of mucociliary clearance through its nonspecific adrenergic activity, phenylephrine's selective α-adrenergic agonism causes vasoconstriction alone, creating a difference in their methods of action.
|
| |
|
| Respiratory system & anti- allergics |
| Anti- Allergics |
Acrivastine
https://en.wikipedia.org/wiki/Acrivastine

Acrivastine is
a medication used for the treatment of allergies and hay fever.
It is a second-generation H1-receptor antagonist antihistamine (like
its base molecule triprolidine)
and works by blocking histamine H1 receptors.Unlike cetirizine or loratadine,
for which the standard dose is one tablet per day, a single acrivastine tablet may be taken up to three times a day. It is not to be taken by people over the age of 65, pregnant women, or people with compromised liver or kidney function.
|
Bilastine
https://en.wikipedia.org/wiki/Bilastine
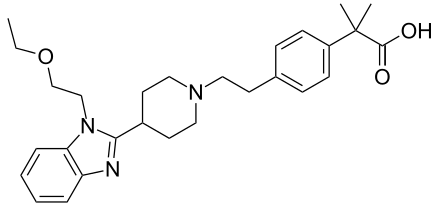
Bilastine, sold under the brand name BILLASI among others, is a second-generation antihistamine medication which is used in the treatment of allergic rhinoconjunctivitis and urticaria (hives).
It exerts its effect as a selective histamine H1 receptor antagonist, and has an effectiveness similar to cetirizine, fexofenadine,
and desloratadine.Bilastine binds to guinea-pig cerebellar histamine H1-receptors (Ki=44 nM) and to human recombinant histamine H1-receptors (Ki=64 nM) with an affinity comparable to that of astemizole and diphenhydramine,
and superior than that of cetirizine by three-fold and fexofenadine by five-fold (Corcóstegui). In different murine models, bilastine by oral route, antagonizes the effects of histamine in a dose-dependent manner, with potency similar to that of cetirizine and between 5.5 and 10 times greater than that of fexofenadine.
|
Buclizine
https://en.wikipedia.org/wiki/Buclizine

Buclizine is
an antihistamine and anticholinergic of
the diphenylmethylpiperazine group.
It is considered to be an antiemetic,
similar to meclizine.
|
Bupivacaine HCl
https://en.wikipedia.org/wiki/Bupivacaine
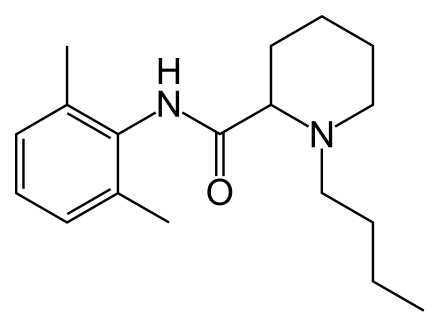
Bupivacaine,
marketed under the brand name Marcaine among
others, is a medication used to decrease feeling in a specific area..Bupivacaine
binds to the intracellular portion of voltage-gated sodium channels and
blocks sodium influx into nerve cells,
which prevents depolarization.
Without depolarization, no initiation or conduction of a pain signal can occur.
|
Cetirizine Dihydrochloride
https://en.wikipedia.org/wiki/Cetirizine

Cetirizine,
sold under the brand name Zyrtec among
others, is a second-generation antihistamine used
to treat allergic rhinitis (hay
fever), dermatitis,
and urticaria.Cetirizine
acts as a highly selective antagonist of
the histamine H1 receptor.[3] The
Ki values for the H1 receptor are approximately
6 nM for cetirizine, 3 nM for levocetirizine, and 100 nM for dextrocetirizine,
indicating that the levorotatory enantiomer is
the main active form. Cetirizine has 600-fold or greater selectivity for the H1 receptor
over a wide variety of other sites, including muscarinic acetylcholine, serotonin, dopamine,
and α-adrenergic receptors, among many others. |
Chlorpheniramine Maleate (Chlorphenamine)
https://en.wikipedia.org/wiki/Chlorphenamine

Chlorphenamine (CP, CPM),
also known as chlorpheniramine, is an antihistamine used
to treat the symptoms of allergic conditions such
as allergic rhinitis (hay
fever).
Chlorphenamine acts primarily as a potent H1 antihistamine. It is specifically a potent inverse agonist of the histamine H1 receptor.The
drug is also commonly described as possessing weak anticholinergic activity by acting as an antagonist of the muscarinic acetylcholine receptors. The dextrorotatory stereoisomer, dexchlorpheniramine,
has been reported to possess Kd values of 15 nM for the H1 receptor and 1,300 nM for the muscarinic acetylcholine receptors in human brain tissue. The smaller the Kd value, the greater the binding affinity of the ligand for its target.
In addition to acting as an inverse agonist at the H1 receptor, chlorphenamine has been found to act as a serotonin reuptake inhibitor (Kd = 15.2 nM for the serotonin transporter).
|
Cloperastine
https://en.wikipedia.org/wiki/Cloperastine

Cloperastine (INN)
or cloperastin, also known as cloperastine
hydrochloride (JAN)
(brand names Hustazol, Nitossil, Seki)
and cloperastine fendizoate (or hybenzoate),
is an antitussive and antihistamine that
is marketed as a cough suppressant in Japan, Hong
Kong, and in some European countries.It
was first introduced in 1972 in Japan, and then in Italy in
1981. The precise mechanism
of action of cloperastine
is not fully clear, but several different biological activities have
been identified for the drug, of which include: ligand of
the σ1 receptor (Ki =
20 nM) (likely an agonist)GIRK
channel blocker (described
as "potent"),antihistamine (Ki =
3.8 nM for the H1 receptor), and anticholinergic. It
is thought that the latter two properties contribute to side effects,
such as sedation and somnolence,
while the former two may be involved in or responsible for the antitussive efficacy of cloperastine. |
Desloratadine
https://en.wikipedia.org/wiki/Desloratadine

Desloratadine (trade
name Clarinex and Aerius)
is a tricyclic H1 antagonist that
is used to treat allergies.
It is an active metabolite of loratadine
Desloratadine is a selective H1-antihistamine which functions as an inverse agonist at the histamine H1 receptor.
At very high doses, is also an antagonist at various subtypes of the muscarinic acetylcholine receptors. This effect is not relevant for the drug's action at therapeutic doses.
|
Fexofenadine
https://en.wikipedia.org/wiki/Fexofenadine

Fexofenadine,
sold under the brand name Allegra among
others, is an antihistamine pharmaceutical
drug used in the treatment
of allergy symptoms, such as hay fever and urticaria. Therapeutically,
fexofenadine is a selective peripheral H1-blocker.
Fexofenadine is a selectively peripheral H1-blocker. Blockage prevents the activation of the H1 receptors by histamine, preventing the symptoms associated with allergies from occurring. Fexofenadine does not readily cross the blood–brain barrier and is therefore less likely to cause drowsiness in comparison to other antihistamines that readily cross the blood-brain barrier (i.e. first-generation antihistamines like diphenhydramine). In general, fexofenadine takes about one hour
to take effect, though this may be affected by the choice of dosage form and the presence/absence of certain foods.
Fexofenadine also exhibits no anticholinergic, antidopaminergic, alpha1-adrenergic, or beta-adrenergic-receptor-blocking effects.
|
Hydroxyzine HCl
https://en.wikipedia.org/wiki/Hydroxyzine

Hydroxyzine,
sold under the brand names Atarax and
others, is a medication of the antihistamine type. It
is used in the treatment of itchiness, anxiety,
and nausea, including
that due to motion sickness.Hydroxyzine's
predominant mechanism of action is
as a potent and selective histamine H1 receptor inverse
agonist.[32][33] This
action is responsible for its antihistamine and sedative effects. Unlike
many other first-generation antihistamines, hydroxyzine has very low affinity for the muscarinic
acetylcholine receptors, and in accordance, has low or no propensity for producing anticholinergic side effects..
|
Levocetirizine HCl
https://en.wikipedia.org/wiki/Levocetirizine

Levocetirizine,
sold under the brand name Xyzal among
others, is an antihistamine used
for the treatment of allergic rhinitis (hay
fever) and long term hives of unclear
cause. It is less sedating than older antihistamines.Levocetirizine is an antihistamine.
It acts as an inverse agonist that decreases activity at histamine
H1 receptors. This in turn prevents the release of other allergy chemicals and increase the blood supply to the area, and provides relief from the typical symptoms of hay fever. |
Loratadine
https://en.wikipedia.org/wiki/Loratadine

Loratadine,
sold under the brand name Claritin among
others, is a medication used to treat allergies. This
includes allergic rhinitis (hay
fever) and hives. It
is also available in combination with pseudoephedrine,
a decongestant,
known as loratadine/pseudoephedrine.Loratadine
is a tricyclic antihistamine, which acts as a selective inverse agonist of
peripheral histamine H1 receptors. The
potency of second generation histamine antagonists is (from strongest to weakest) desloratadine (Ki 0.4 nM)
> levocetirizine (Ki 3 nM)
> cetirizine (Ki 6 nM) > fexofenadine (Ki 10 nM)
> terfenadine > loratadine. However, the onset of action varies significantly and clinical efficacy is not always directly related to only the H1 receptor potency, as concentration of free drug at the receptor must also be considered.
|
Rupatadine Fumarate
https://en.wikipedia.org/wiki/Rupatadine

Rupatadine is
a second generation antihistamine and PAF antagonist
used to treat allergies.
Rupatadine is a second generation, non-sedating, long-acting histamine antagonist with selective peripheral H1 receptor antagonist activity. It further blocks the receptors of the platelet-activating factor (PAF) according to in vitro and in vivo studies.
Rupatadine possesses anti-allergic properties such as the inhibition of the degranulation of mast cells induced by immunological and non-immunological stimuli, and inhibition of the release of cytokines, particularly of the tumor necrosis factors (TNF) in human mast cells and monocytes.
|
Triprolidine HCl
https://en.wikipedia.org/wiki/Triprolidine
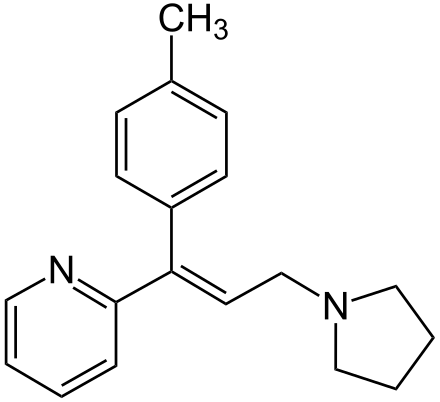
riprolidine is
an over-the-counter antihistamine with anticholinergic properties. It
is used to combat the symptoms associated with allergies and
is sometimes combined with other cold medications
designed to provide general relief for flu-like
symptoms. As with many
antihistamines, the most common side effect is
drowsiness. |
| Anti- Asthma/COPD |
Acebrophylline
https://pharmeasy.in/molecules/acebrophylline-8210#:~:text=Acebrophylline%20is%20a%20Xanthine%20derivative,read%20more

Acebrophylline is
a Xanthine derivative. It is used in obstructive airway diseases such as asthma and COPD (Chronic Obstructive Pulmonary Disease) as a bronchodilator to improve difficulty in breathing.
|
Ciclesonide
https://en.wikipedia.org/wiki/Ciclesonide

Ciclesonide is
a glucocorticoid used
to treat asthma and allergic
rhinitis
Ciclesonide is a glucocorticoid used to treat asthma and allergic rhinitis. It is marketed under the brand names Alvesco for asthma and Omnaris, Omniair, Zetonna, and Alvesco for hay fever in the US and Canada.
Side effects of the medication include headache, nosebleeds, and inflammation of the nose and throat linings.
|
Ipratropium Bromide
https://en.wikipedia.org/wiki/Ipratropium_bromide

Ipratropium bromide,
sold under the trade name Atrovent among
others, is a medication which opens up the medium and large airways in the lungs. It
is used to treat the symptoms of chronic obstructive pulmonary disease and asthma. It
is used by inhaler or nebulizer.
Chemically, ipratropium bromide is a quaternary ammonium compound (which is indicated by the -ium per the BAN and the USAN) obtained by treating atropine with isopropyl
bromide, thus the name: isopropyl + atropine.[citation needed] It is chemically related to components of the plant Datura
stramonium, which was used in ancient India for asthma.
Ipratropium exhibits broncholytic action by reducing cholinergic influence on the bronchial musculature. It blocks muscarinic acetylcholine receptors, without specificity for subtypes, and therefore promotes the degradation of cyclic guanosine monophosphate (cGMP), resulting in a decreased intracellular concentration of cGMP. Most likely due to actions of cGMP on intracellular calcium, this results in decreased contractility of smooth muscle in the lung, inhibiting bronchoconstriction and mucus secretion.
It is a nonselective muscarinic antagonist, and does not diffuse into the blood, which prevents systemic side effects. Ipratropium is a derivative of atropine but is a quaternary
amine and therefore does not cross the blood–brain barrier, which prevents central side effects (anticholinergic syndrome). Ipratropium should never be used in place of salbutamol (albuterol) as a rescue medication.
|
Ozagrel HCl
https://en.wikipedia.org/wiki/Ozagrel

Ozagrel (INN)
is an antiplatelet agent working
as a thromboxane A2 synthesis
inhibitor.
|
Roflumilast
https://en.wikipedia.org/wiki/Roflumilast

Roflumilast,
sold under the trade name Daxas among
others, is a drug that acts as a selective, long-acting inhibitor of the enzyme phosphodiesterase-4 (PDE-4).
It has anti-inflammatory effects
and is used as an orally administered drug for the treatment of inflammatory conditions of the lungs such as chronic obstructive pulmonary disease (COPD).Its
primary clinical use is in the prevention of exacerbations (lung attacks) in severe chronic obstructive pulmonary disease (COPD).
|
Tiotropium Bromide / Monohydrate
https://en.wikipedia.org/wiki/Tiotropium_bromide

Tiotropium bromide,
sold under the brand name Spiriva among
others, is a long-acting bronchodilator used
in the management of chronic obstructive pulmonary disease (COPD)
and asthma. Specifically
it is used to try to prevent periods of worsening rather than for those periods themselves.Tiotropium is a muscarinic receptor antagonist,
often referred to as an antimuscarinic or anticholinergic agent. Although it does not display selectivity for specific muscarinic receptors, when topically applied it acts mainly on M3 muscarinic
receptors located on smooth muscle cells and submucosal glands. This leads to a reduction in smooth muscle contraction and mucus secretion and thus produces a bronchodilatory effect.
|
Erdosteine
https://en.wikipedia.org/wiki/Erdosteine

Erdosteine is
a molecule with mucolytic activity.
Structurally is a thiol derivative
characterized by the presence of two thiol groups. These
two functional sulfhydryl groups
contained in the molecule are released following first-pass metabolism with
the conversion of erdosteine into its pharmacologically active metabolite Met-I.
|
Terbutaline Sulphate
https://en.wikipedia.org/wiki/Terbutaline

Terbutaline, sold under the brand name Bricanyl among
others, is a β2 adrenergic receptor agonist,
used as a "reliever" inhaler in the management of asthma symptoms
and as a tocolytic (anti-contraction medication)
to delay preterm labor for
up to 48 hours. This time can then be used to administer steroid injections to the mother which help fetal lung maturity and reduce complications of prematurity.[1] It
should not be used to prevent preterm labor or delay labor more than 48–72 hours.The tertiary butyl group in terbutaline makes it more selective for β2 receptors.
Since there is no hydroxy group on position 4 of the benzene ring, the molecule is less susceptible to metabolism by the enzyme catechol-O-methyl transferase.
|
| Bronchodilators & Mast Cell Stabilizers |
Montelukast Sodium
https://en.wikipedia.org/wiki/Montelukast

Montelukast,
sold under the brand name Singulair among
others, is a medication used in the maintenance treatment of asthma.
Montelukast is in the leukotriene receptor antagonist family of medications. It works by blocking the action of leukotriene D4 in the lungs resulting in decreased inflammation and relaxation of smooth
muscle.
Montelukast functions as a leukotriene receptor antagonist (cysteinyl leukotriene receptors) and consequently opposes the function of these inflammatory mediators; leukotrienes are produced by the immune system and serve to promote bronchoconstriction,
inflammation, microvascular permeability, and mucus secretion in asthma and COPD. Leukotriene receptor antagonists are sometimes colloquially referred to as leukasts.
Two genes of interest are ALOX5 and LTC4S, which catalyze two major steps in the biosynthetic pathway of leukotrienes.
|
| |
|
| Supplementation |
| Appetite Suppressant |
Phentermine HCl
https://en.wikipedia.org/wiki/Phentermine

Phentermine (phenyl-tertiary-butylamine),
sold under the brand name Ionamin among
others, is a medication used together with diet and exercise to treat obesity. It
is taken by mouth for up to a few weeks. After
a few weeks the beneficial effects no longer occur. It
is also available as the combination phentermine/topiramate.Phentermine
has some similarity in its pharmacodynamics with
its parent compound, amphetamine, as they both are TAAR1
agonists, where the activation of TAAR1 in monoamine neurons
facilitates the efflux, or release into the synapse, of these neurochemicals.
|
| Dietary Supplement |
Nicotinamide
https://en.wikipedia.org/wiki/Nicotinamide

Nicotinamide (NAM)
is a form of vitamin B3 found
in food and used as a dietary supplement and
medication.As a supplement, it is used by mouth to prevent and treat pellagra (niacin
deficiency).While nicotinic acid (niacin)
may be used for this purpose, nicotinamide has the benefit of not causing skin flushing. As
a cream, it is used to treat acne.Side
effects are minimal.At high doses liver problems may occur. Normal amounts are safe for use during pregnancy. Nicotinamide
is in the vitamin B family of medications, specifically the vitamin
B3 complex. It
is an amide of nicotinic acid. Foods that contain nicotinamide include yeast, meat,
milk, and green vegetables.
|
| Phosphorus Supplementation |
Butaphosphan
https://www.trc-canada.com/product-detail/?CatNum=B689410

Butafosfan, a component of Catosal, is licensed for the treatment of metabolic disorders or as a tonic in swine. It reduces the stress-induced cortisol (C696302, d4 labelled) response after mixing of unfamiliar pigs. It can also increase milk production in Holstein cows in combination with cyanocobalamin.
|
| |
|
| Surgical & Vaccines |
| Intravenous & Other Sterile Solutions |
Hydroxyethyl Starch
https://en.wikipedia.org/wiki/Hydroxyethyl_starch

Hydroxyethyl starch (HES/HAES), sold under the brand name Voluven among others, is a nonionic starch derivative, used as a volume expander in intravenous therapy. The use of HES on critically ill patients is associated with an increased risk of death and kidney problems.HES is a general term and can be sub-classified according to average molecular weight, molar substitution, concentration, C2/C6 ratio and Maximum Daily Dose.
|
Maize Starch
https://en.wikipedia.org/wiki/Corn_starch

Corn starch, maize starch, or corn flour (British English) is the starch derived from corn (maize) grain. The starch is obtained from the endosperm of the kernel. Corn starch is a common food ingredient, often used to thicken sauces or soups, and to make corn syrup and other sugars Corn
starch is versatile, easily modified, and finds many uses in industry such as adhesives, in paper products, as an anti-sticking agent, and textile manufacturing It has medical uses as well, such as to supply glucose for people with glycogen storage disease
Like many products in dust form, it can be hazardous in large quantities due to its flammability. When mixed with a fluid, corn starch can rearrange itself into a non-Newtonian fluid. For example, adding water transforms corn starch into a material commonly known as oobleck while adding oil transforms corn starch into an electrorheological
(ER) fluid. The concept can be explained through the mixture termed "cornflour slime".
Corn starch is the preferred anti-stick agent on medical products made from natural latex, including condoms, diaphragms, and medical gloves.
Corn starch has properties enabling supply of glucose to maintain blood sugar levels for people with glycogen storage disease. Corn starch can be used starting at age 6–12 months allowing glucose fluctuations to be deterred.
|
| Mucolytics, Proteolytic & Other Enzymes |
Serratiopeptidase
https://en.wikipedia.org/wiki/Serratiopeptidase

Serratiopeptidase (Serratia
E-15 protease, also known as serralysin, serrapeptase, serratiapeptase, serratia
peptidase, serratio
peptidase, or serrapeptidase),
is a proteolytic enzyme (protease) produced
by non-pathogenic enterobacterium Serratia sp.
E-15, now known as Serratia marcescens ATCC
21074. This
microorganism was originally isolated in the late 1960s from silkworm Bombyx
mori L. (intestine), Serratiopeptidase
is present in the silkworm intestine and allows the emerging moth to dissolve its cocoon. Serratiopeptase is produced by purification from culture of Serratia E-15 bacteria. It is a member of the Peptitase M10B (Matrixin)
family.
|
Trypsin And Chymotrypsin
https://en.wikipedia.org/wiki/Trypsin
https://en.wikipedia.org/wiki/Chymotrypsin

Trypsin (EC 3.4.21.4)
is a serine protease from
the PA clan superfamily,
found in the digestive system of
many vertebrates,
where it hydrolyzes proteins. Trypsin
is formed in the small intestine when
its proenzyme form,
the trypsinogen produced
by the pancreas, is
activated. Trypsin cuts peptide chains
mainly at the carboxyl side
of the amino acids lysine or arginine.
It is used for numerous biotechnological processes.
The process is commonly referred to as trypsin proteolysis or trypsinisation,
and proteins that have been digested/treated with trypsin are said to have been trypsinized.Chymotrypsin (EC 3.4.21.1,
chymotrypsins A and B, alpha-chymar ophth, avazyme, chymar, chymotest, enzeon, quimar, quimotrase, alpha-chymar, alpha-chymotrypsin A, alpha-chymotrypsin) is a digestive enzyme component of pancreatic
juice acting in the duodenum, where it performs proteolysis,
the breakdown of proteins and polypeptides.
|
| Non-Ionic Iodinated |
Iopamidol
https://en.wikipedia.org/wiki/Iopamidol

Iopamidol (INN),
sold under the brand name Isovue among
others, is a nonionic, low-osmolar iodinated contrast agent,
developed by Bracco Diagnostics.Iopamidol
is indicated for angiography throughout the cardiovascular systemIt is also indicated for intrathecal administration in adult neuroradiology including myelography (lumbar, thoracic, cervical, total columnar), and for contrast enhancement of computed tomographic (CECT) cisternography and ventriculography. Isovue-M 200 (lopamidol Injection) is indicated for thoraco-lumbar myelography in children over the age of two years.
|
Clenbuterol HCl
https://en.wikipedia.org/wiki/Clenbuterol

Clenbuterol is
a sympathomimetic amine used
by sufferers of breathing disorders as a decongestant and bronchodilator.
People with chronic breathing disorders such as asthma use
this as a bronchodilator to make breathing easier. It is most commonly available as the hydrochloride salt,
clenbuterol hydrochloride.
Clenbuterol is approved for use in some countries as a bronchodilator for asthma.
Clenbuterol is a β2 agonist with some structural and pharmacological similarities to epinephrine and salbutamol, but its effects are more potent and longer-lasting as a stimulant and thermogenic drug.[ It
is commonly used for smooth muscle-relaxant properties as a bronchodilator and tocolytic.
It is classified by the World Anti-Doping Association as an anabolic agent, not as a β2 agonist.
Clenbuterol is prescribed for treatment of respiratory diseases for horses, and as an obstetrical aid in cattle. It is illegal in some countries to use in livestock used for food.
|
Cyclobenzaprine HCl
https://en.wikipedia.org/wiki/Cyclobenzaprine

Cyclobenzaprine,
sold under the brand name Flexeril among
others, is a medication used for muscle spasms from
musculoskeletal conditions of sudden onset. It
is not useful in cerebral palsy.Cyclobenzaprine
is a centrally acting muscle relaxant. Cyclobenzaprine
is a 5-HT2 receptor antagonist; it relieves muscle spasm through action on the central nervous system at the brain stem, rather than targeting the peripheral nervous system or muscles themselves.
|
Vecuronium Bromide
https://en.wikipedia.org/wiki/Vecuronium_bromide

Vecuronium bromide,
sold under the brand name Norcuron among
others, is a medication used as part of general anesthesia to
provide skeletal muscle relaxation during surgery or mechanical
ventilation.Vecuronium operates by competing for the cholinoceptors at the motor end plate, thereby exerting its muscle-relaxing properties
|
| Peripherally Acting Muscle Relaxants |
Lidocaine (Lignocaine)
ttps://en.wikipedia.org/wiki/Lidocaine
Lidocaine,
also known as lignocaine, is a medication used to numb
tissue in a specific area (local
anesthetic). It is also
used to treat ventricular tachycardia and
to perform nerve blocks.
Lidocaine alters signal conduction in neurons by prolonging the inactivation of the fast voltage-gated Na+ channels in the neuronal cell membrane responsible for action potential propagation. With sufficient blockage, the
voltage-gated sodium channels will not open and an action potential will not be generated. Careful titration allows for a high degree of selectivity in the blockage of sensory neurons, whereas higher concentrations also affect other types of neurons.
The same principle applies for this drug's actions in the heart. Blocking sodium channels in the conduction system, as well as the muscle cells of the heart, raises the depolarization threshold, making the heart less likely to initiate or conduct early action potentials that may cause an arrhythmia.
|
Myrtecaine
ttps://en.wikipedia.org/wiki/Myrtecaine

Myrtecaine (Nopoxamine),
sold as a combination product with diethylamine salicylate under
the trade name Algesal and Algésal
Suractivé among others, is
a local anaesthetic in
the form of a topical cream, or
with laurilsulfate in rubefacient preparations. It
is used to treat muscle strains, tendinitis or ligament sprains and
joint pain. It is a
surface anaesthetic, adds to the analgesic and
anti-inflammatory actions of diethylamine salicylate by facilitating its penetration. Also
myrtecaine has a muscle relaxant effect. |
| |
|
| Veterinary list |
| Veterinary |
Albendazole
https://en.wikipedia.org/wiki/Albendazole
Albendazole,
also known as albendazolum, is
a medication used for the treatment of a variety of parasitic worm infestations.
As a vermicide, albendazole causes degenerative alterations in the intestinal cells of the worm by binding to the colchicine-sensitive site of β-tubulin, thus inhibiting its polymerization or assembly into microtubules (it binds much better to the β-tubulin of parasites
than that of mammals). Albendazole leads to impaired uptake of glucose by the larval and adult stages of the susceptible parasites, and depletes their glycogen stores. Albendazole also prevents the formation of spindle fibers needed for cell division, which in turn blocks egg production and development; existing eggs are prevented from hatching. Cell motility, maintenance of cell shape, and intracellular transport are also disrupted.At higher concentrations, it disrupts the helminths' metabolic pathways by inhibiting metabolic enzymes such as malate dehydrogenase and fumarate
reductase, with inhibition of the latter leading to less energy produced by the Krebs cycle. Due to diminished ATP production, the parasite is immobilized and eventually dies.
Some parasites have evolved to have some resistance to albendazole by having a different set of acids comprising β-tubulin, decreasing the binding affinity of albendazole. Drosophilia have many of the same mutations, meaning the drug does not affect fruit flies.
|
Buparvaquone
https://en.wikipedia.org/wiki/Buparvaquone

Buparvaquone is
a hydroxynaphthoquinone antiprotozoal drug
related to parvaquone and atovaquone.
It is a promising compound for the therapy and prophylaxis of all forms of theileriosis.
Buparvaquone has been shown to have anti-leishmanial activity in
vitro. It can be used to treat bovine East
Coast fever protozoa in
vitro, along with the only other substance known – Peganum
harmala.[ It
is the only really effective commercial therapeutic product against bovine theileriosis,
where it has been used since the late 1980s.
|
Butaphosphan
https://www.trc-canada.com/product-detail/?CatNum=B689410
Butafosfan, a component of Catosal, is licensed for the treatment of metabolic disorders or as a tonic in swine. It reduces the stress-induced cortisol (C696302, d4 labelled) response after mixing of unfamiliar pigs. It can also increase milk production in Holstein cows in combination with cyanocobalamin.
|
Clorsulon
ttps://www.trc-canada.com/product-detail/?C587380

A benzenedisulfonamide derivative with fasciolicidal activity. Anthelmintic (Trematodes).
|
Diminazene Diaceturate
https://en.wikipedia.org/wiki/Diminazene
Diminazene (INN; also known as diminazen) is an anti-infective medication for animals that is sold under a variety of brand names. It is effective against certain protozoa such as Babesia, Trypanosoma,
and Cytauxzoon. The drug may also be effective against certain bacteria including Brucella and Streptococcus.
Chemically it is a di-amidine and it is formulated as its aceturate salt, diminazene aceturate.
The mechanism is not well understood; it probably inhibits DNA replication, but also has affinity to RNA.
|
Quinapyramine Chloride / Sulphate
https://en.wikipedia.org/wiki/Quinapyramine

Quinapyramine is
a trypanocidal agent for
veterinary use.
|
Triclabendazole
https://en.wikipedia.org/wiki/Triclabendazole

Triclabendazole,
sold under the brand name Egaten among
others, is a medication used to treat fascioliasis and paragonimiasis. It
is very effective for both conditions. Treatment
in hospital may be required..It is a member of the benzimidazole family
of anthelmintics.
The benzimidazole drugs share a common molecular structure, triclabendazole being the exception in having a chlorinated benzene ring but no carbamate group.
Benzimidazoles such as triclabendazole are generally accepted to bind to beta-tubulin therefore
preventing the polymerization of microtubules.
|
| |
|
| We manufacture |
| |
Adenosylcobalamin
https://en.wikipedia.org/wiki/Adenosylcobalamin

.
Adenosylcobalamin (AdoCbl), also known as coenzyme B12, cobamamide, and dibencozide, is, along with methylcobalamin (MeCbl), one of the biologically active forms of vitamin B12.
Adenosylcobalamin participates as a cofactor in radical-mediated 1,2-carbon skeleton rearrangements. These processes require the formation of the deoxyadenosyl radical through homolytic dissociation of the carbon-cobalt bond. This bond is exceptionally weak, with a bond dissociation energy of 31 kcal/mol, which is further lowered in the chemical environment of an enzyme active site. An enzyme that uses adenosylcobalamin as a cofactor is methylmalonyl-CoA
mutase (MCM).
|
| |
Beclomethasone Dipropionate
https://en.wikipedia.org/wiki/Beclometasone

Beclometasone,
also known as beclometasone dipropionate, and sold under
the brand name Qvar among
others, is a steroid medication. It
is available as an inhaler,
cream, pills, and nasal spray. The
inhaled form is used in the long-term management of asthma. The
cream may be used for dermatitis and psoriasis. The
pills have been used to treat ulcerative colitis. The
nasal spray is used to treat allergic rhinitis and nasal
polyps.
Beclometasone is mainly a glucocorticoid. Glucocorticoids are corticosteroids that bind to the glucocorticoid receptor that is present in almost every vertebrate animal cell. The activated glucocorticoid receptor-glucocorticoid complex up-regulates the expression of anti-inflammatory proteins in the nucleus (a
process known as transactivation) and represses the expression of proinflammatory proteins in the cytosol by preventing the translocation of other transcription factors from the cytosol into the nucleus (transrepression).
Glucocorticoids are part of the feedback mechanism in the immune system which reduces certain aspects of immune function, such as inflammation.
|
| |
Betamethasone Valerate
https://en.wikipedia.org/wiki/Betamethasone_valerate

Betamethasone valerate is
a synthetic glucocorticoid ester.
It is the 17-valerate ester
of betamethasone.
|
| |
Betamethasone Dipropionate
ttps://en.wikipedia.org/wiki/Betamethasone_dipropionate

Betamethasone dipropionate is a glucocorticoid steroid with anti-inflammatory and immunosuppressive abilities. It is applied as a topical cream,
ointment, lotion or gel (Diprolene) to treat itching and other minor skin conditions such as eczema.
Minor side effects include dry skin and mild, temporary stinging when applied.
Betamethasone dipropionate is a "super high potency" corticosteroid used to treat inflammatory skin conditions such as dermatitis, eczema and psoriasis. It is a synthetic analog of the adrenal corticosteroids. Although its exact mechanism of action is not known, it is effective when applied topically to cortico-responsive inflammatory dermatoses.
|
| |
Betamethasone Sodium Phosphate
https://en.wikipedia.org/wiki/Betamethasone_phosphate

Betamethasone sodium phosphate is
a synthetic glucocorticoid corticosteroid and
a corticosteroid ester.
|
| |
Clobetasol Propionate
https://en.wikipedia.org/wiki/Clobetasol_propionate

Clobetasol propionate is
a corticosteroid used
to treat skin conditions such as eczema, contact
dermatitis, seborrheic
dermatitis, and psoriasis. It
is applied to the skin as a cream, ointment, or shampoo. Use
should be short term and only if other weaker corticosteroids are not effective. Use
is not recommended in rosacea or perioral
dermatitis. |
| |
Deflazacort
https://en.wikipedia.org/wiki/Deflazacort
Deflazacort (trade
name Calcort among
others) is a glucocorticoid used
as an anti-inflammatory and immunosuppressant.
Deflazacort is an inactive prodrug which is metabolized rapidly to the active drug 21-desacetyldeflazacort.
|
| |
Diacerein
https://en.wikipedia.org/wiki/Diacerein
Diacerein (INN),
also known as diacetylrhein, is a slow-acting medicine of
the class anthraquinone used
to treat joint diseases such as osteoarthritis (swelling and pain in the joints).Diacerein works by blocking the actions of interleukin-1 beta, a protein involved in the inflammation and destruction of cartilage that
play a role in the development of symptoms of degenerative joint diseases such as osteoarthritis. Due to its specific mode of action, which does not involve the inhibition of prostaglandin synthesis,
diacerein has been shown to have anti-osteoarthritis and cartilage stimulating properties in vitro and animal models. |
| |
Hydrocortisone Acetate
https://en.wikipedia.org/wiki/Hydrocortisone_acetate

Hydrocortisone acetate is
a synthetic glucocorticoid corticosteroid and
a corticosteroid ester.
|
| |
Hydroxycobalamin
https://en.wikipedia.org/wiki/Hydroxocobalamin

Hydroxocobalamin,
also known as vitamin B12a and hydroxycobalamin,
is a vitamin found
in food and used as a dietary supplement. As
a supplement it is used to treat vitamin B12 deficiency including pernicious
anemia. Other
uses include treatment for cyanide poisoning, Leber's
optic atrophy, and toxic
amblyopia.
Vitamin B12 refers to a group of compounds called cobalamins that are available in the human body in a variety of mostly interconvertible forms. Together with folate, cobalamins are essential cofactors required for DNA synthesis in cells where chromosomal replication and division are occurring—most notably the bone
marrow and myeloid cells. As a cofactor, cobalamins are essential for two cellular reactions:
|
| |
Methyl Prednisolone Acetate

Methylprednisolone acetate,
sold under the brand names Depo-Medrol among
others, is a synthetic glucocorticoid corticosteroid and
a corticosteroid ester—specifically
the C21 acetate ester of methylprednisolone—which
is used in clinical and veterinary
medicine. It
has been formulated as an aqueous suspension for intramuscular, intra-articular, soft
tissue, and intralesional injection alone
and in combination with lidocaine,
a local anesthetic. Methylprednisolone
acetate was previously suspended with polyethylene glycol but
is no longer formulated with this excipient due
to concerns about possible toxicity.
|
| |
Methyl Prednisolone Base
https://en.wikipedia.org/wiki/Methylprednisolone
Methylprednisolone,
sold under the brand name Medrol among
others, is a corticosteroid medication
used to suppress the immune system and
decrease inflammation. Conditions
in which it is used include skin diseases, rheumatic disorders,
allergies, asthma, croup, COPD,
certain cancers, multiple
sclerosis, and as add-on therapy for tuberculosis or
radiculopathyUnbound glucocorticoids cross cell membranes and bind with high affinity to specific cytoplasmic receptors, modifying transcription and protein synthesis. By this mechanism, glucocorticoids can inhibit leukocyte infiltration at the site of inflammation, interfere with mediators of inflammatory response, and suppress humoral immune responses. The anti-inflammatory actions of corticosteroids are thought to involve phospholipase A2 inhibitory proteins, lipocortins, which control the
biosynthesis of potent mediators of inflammation such as prostaglandins and leukotrienes.
|
|