|
GC-Depth, GC-Content
Distribution
In molecular biology and genetics, GC-content (or guanine-cytosine content)
is the percentage of nitrogenous baseson
a DNA or RNA molecule
that are either guanine or cytosine (from
a possibility of four different ones, also including adenine and thymine in
DNA and adenine and uracil in
RNA).[1] This
may refer to a certain fragment of DNA or RNA, or that of the whole genome.
When it refers to a fragment of the genetic material, it may denote the
GC-content of section of a gene (domain), single gene, group of genes (or gene
clusters), or even a non-coding region.
Genomic content
Within-genome variation
The GC ratio within a genome is found to be markedly variable. These variations
in GC ratio within the genomes of more complex organisms result in a mosaic-like
formation with islet regions called isochores.[11] This
results in the variations in staining intensity in the chromosomes.[12] GC-rich
isochores include in them many protein coding genes, and thus determination of
ratio of these specific regions contributes in mapping gene-rich regions of the
genome.[13][14]
Coding sequences
Within a long region of genomic sequence, genes are often
characterised by having a higher GC-content in contrast to the background
GC-content for the entire genome. Evidence of GC ratio with that of length of
the coding region of a gene has shown that the length of the coding
sequence is directly proportional to higher G+C content.[15] This has been
pointed to the fact that the stop codon has a bias towards A and T
nucleotides, and, thus, the shorter the sequence the higher the AT bias.[16]
Comparison of more than 1,000 orthologous genes in mammals showed
marked within-genome variations of the third-codon position GC content, with a range
from less than 30% to more than 80%.[17]
Among-genome variation
GC content is found to be variable with different
organisms, the process of which is envisaged to be contributed to by variation
in selection, mutational bias, and
biased recombination-associated DNA repair.[18]
|

Nucleotide bonds showing AT and GC pairs. Arrows point to the hydrogen bonds.
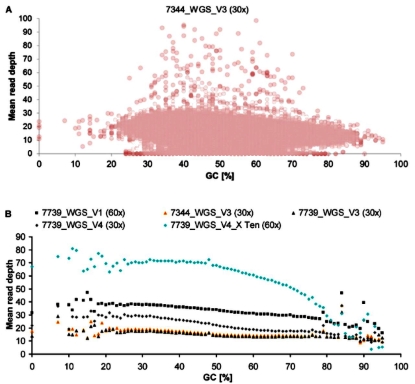
Influence of GC Content on Mean Read Depth in WGS |
|
The average GC-content in human genomes ranges from 35%
to 60% across 100-Kb fragments, with a mean of 46.1%.[17] The GC-content
of Yeast (Saccharomyces cerevisiae) is 38%,[19] and that of
another common model organism, thale cress (Arabidopsis thaliana), is 36%.[20] Because of the
nature of the genetic code, it is virtually impossible for an
organism to have a genome with a GC-content approaching either 0% or 100%.
However, a species with an extremely low GC-content is Plasmodium falciparum (GC%
= ~20%),[21] and it is
usually common to refer to such examples as being AT-rich instead of GC-poor.[22]
Several mammalian species (e.g., shrew, microbat, tenrec, rabbit) have independently undergone a marked increase in
the GC-content of their genes. These GC-content changes are correlated with
species life-history traits (e.g., body
mass or longevity) and genome size,[17] and might be
linked to a molecular phenomenon called the GC-biased gene conversion.[23]
|